





Background
Fungal secondary metabolites are chemically diverse, and some display pharmaceutical potential as inhibitors of inflammation, cytotoxicity, and cancer (Shao et al. 2011; Wei et al. 2010). Gliotoxin, a member of the epipolythiodioxopiperazine family of secondary metabolites, is characterized by a disulfide bond across a piperazine ring. It has been isolated from several species of fungi, including the human pathogen Aspergillus (Waring et al. 1995). Gliotoxin is known to possess immunosuppressive properties, and it causes apoptosis in immune cells including neutrophils, eosinophils, macrophages, and thymocytes (Sutton et al. 1994; Pahl et al. 1996; Lopez-Franco et al. 2002). Gliotoxin’s immunosuppressive properties result from its inhibition of nuclear factor kappa B (NF-κB) because it represses proteasome-mediated degradation of the NF-κB antagonist, IκB-α (Kroll et al. 1999; Lopez-Franco et al. 2002). Gliotoxin also enters the redox cycle and transitions between reduced and oxidized forms after uptake into cells. Reactive oxygen species (ROS) are produced as a by-product, and this induces DNA damage and apoptosis in immune cells (Harms et al. 2015). ROS have also been reported to inhibit NF-κB through direct oxidation (Toledano and Leonard 1991; Nowak et al. 2008). In this regard, the role of gliotoxin in regulating NF-κB and ROS has focused on immunological therapeutic applications. However, recent publications suggest that gliotoxin has antitumor properties (Kweon et al. 2003; Bhatnagar and Kim 2015), although the mechanistic details remain unclear. In the present study, we investigated the antiproliferative effects of gliotoxin on HT1080 human fibrosarcoma cells. We found that gliotoxin from the marine fungus Aspergillus fumigatu regulates proliferation of HT1080 cells by inducing apoptosis. This cytotoxicity was related to the inactivation of NF-κB and an increase in intracellular ROS.
Methods
Gliotoxin from the marine fungus Aspergillus fumigatus was obtained as described by Bhatnagar and Kim (2015). Briefly, A. fumigatus was isolated from the surface of marine green algae, which were collected in Cheongsapo, Busan, Republic of Korea. The fungal strain was cultured in YPGA media containing 0.1 % yeast extract, 2 % peptone, 1 % glucose, 2 % agar, 60 % seawater, and 40 % distilled water. The fermentation broth was extracted (2.3 g) with ethyl acetate (EtOAc, 1:1.5 v/v; broth-EtOAc, 1:1 v/v). The extract was fractionated by silica gel chromatography (n-hexane-EtOAc, 100 %:0 %; CH2Cl2-methyl alcohol (MeOH), 1:1), ODS column chromatography (H2O-MeOH, 100 %:100 %), and Sephadex LH-20 column chromatography (H2O-MeOH, 100 %:100 %). High-performance liquid chromatography (HPLC) using a YMC ODS-A column (250 mm × 10 mm I.D., S-5 μm, 12 nm, MeOH) yielded purified gliotoxin (5.2 mg). The structure and molecular formula of gliotoxin was ascertained using 1H and 13C nuclear magnetic resonance (NMR), as well as low-resolution electron impact mass spectrometry (LREI-MS) data. 1H NMR spectrum (DMSO-d6, 400 MHz): 3.44 (1H, dd, J = 4.8, H-3a), 4.28 (1H, dd, J = 9.9, H-3a), 4.39 (1H, dd, J = 6.8, H-5), 4.842 (1H, m, H-6), 5.78 (1H, d, J = 9.9, H-7), 5.95 (1H, m, H-8), 6.00 (1H, m, H-9), 2.96, 3,73 (1H, d, J = 18.1, H-10), 3.20 (3H, s, H-11). 13C NMR spectrum (DMSO-d6, 100 MHz): 166.0 (C-1), 77.2 (C-3), 60.5 (C-3), 165.2 (C-4), 69.8 (C-5), 75.6 (C-6), 129.9 (C-7), 123.4 (C-8), 120.2 (C-9), 130.7 (C-19a), 36.6 (C-10), 73.1 (C-10a), 27.5 (C-11). LREI-MS m/z: 349 [M]+ (C21H23N3O2). LREI-MS m/z: 326.38 [M]+ (C13H14N2O4 S2).
Human fibrosarcoma cells (HT1080) were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). Cells were cultured in DMEM supplemented with penicillin/streptomycin and fetal bovine serum (FBS) (Gibco BRL, Life Technology, NY, USA). Cytotoxicity assays were performed with 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma Chemical Co., St. Louis, MO, USA). All antibodies used for western blot analysis were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Other chemicals and reagents were of commercially available analytical grade.
HT1080 cells were cultured on DMEM (Gibco, USA) containing 10 % FBS and incubated at 37 °C under a humidified atmosphere of 5 % CO2. Cytotoxicity of gliotoxin on HT1080 cells was analyzed using an MTT assay. To estimate the cytotoxicity of gliotoxin, cells were seeded in a 96-well plate at a concentration of 4 × 104 cells/ml and incubated for 24 h. The cells were then exposed to different concentrations (0, 25, 50, or 100 μM) of gliotoxin at 37 °C under a humidified atmosphere of 5 % CO2. Subsequently, 100 μl MTT solution (1 mg/ml) was added to each well, and cells were incubated for another 3 h. The formazan salt that formed was dissolved in 100 μl DMSO. The absorbance was measured at 550 nm (GENiosr Microplate Reader, Tecan Austria GmbH, Austria).
Apoptotic cells were stained using the fluorescence isothiocyanate (FITC) Annexin V apoptosis detection kit (BD Biosciences) according to the manufacturer’s instructions. Briefly, HT1080 cells were cultured in 6-well plates at a density of 5 × 104 cells/ml. The cells were then treated with 100 μM gliotoxin for 24 h. After incubation, trypsinized, floating, and adherent cells were pooled and centrifuged. Harvested cells were washed with PBS twice, mixed in 1× binding buffer, and incubated with Annexin V/PI double-staining solution at room temperature (RT) for 20 min. The stained cells were analyzed by flow cytometry (FACSCalibur, BD Science, Heidelberg, Germany), and the percentage of apoptotic cells was calculated using Cell Quest software.
After the indicated incubation times with or without 100 μM gliotoxin, HT1080 cells were lysed. Nuclear and cytoplasmic extracts were prepared according to Duvoix et al. (2004). Briefly, cell pellets were suspended in ice-cold hypotonic lysis buffer containing protease inhibitors. We added 10 % Igepal (Sigma-Aldrich) to the cell suspension and vortexed for 10 s to lyse the cells. The lysate was then centrifuged at 18,000 rpm for 1 min. The cytoplasmic supernatant was stored, and cell pellets were washed with hypotonic lysis buffer. Nuclear extraction buffer was added to each pellet, and the cell suspension was gently mixed on an orbital shaker for 15 min at 4 °C. This was followed by centrifugation at 10,500 rpm for 7 min at 4 °C. Finally, the nuclear supernatant was transferred to a prechilled microcentrifuge tube and stored at −80 °C.
Western blotting was performed according to the standard procedures. Briefly, cells were cultured at a density of 5 × 104 cells/ml in six-well plates with serum-free media. After incubation for 24 h, cells were treated with gliotoxin for another 24 h. Cells were lysed in RIPA (150 mM NaCl, 1.0 % IGEPAL® 0.5 % sodium deoxycholate, 0.1 % sodium dodecylsulfate, 50 mM Tris, pH 8.0) buffer at 4 °C for 30 min. Proteins from nuclear and cytoplasmic extracts were obtained, and 100 μg/ml of protein was separated using 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis with a 5 % stacking gel. Separated proteins were transferred onto a nitrocellulose membrane (Amersham Pharmacia Biotech, England, UK). The membrane was blocked for 1.5 h at RT using Tris-buffered saline and 0.1 % Tween-20 (TBS-T) with 5 % skim milk. After washing the membrane with TBS-T twice, the blots were incubated for 1 h with the indicated antibodies at 25 °C. The respective proteins were detected with a chemiluminescent ECL assay kit (Amersham Pharmacia, England, UK) according to the manufacturer’s instructions.
Results
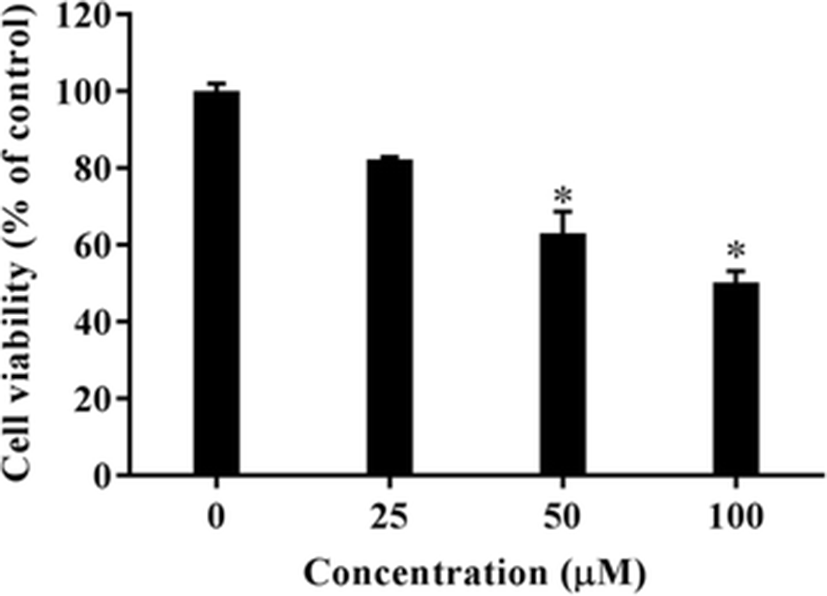
To examine the antiproliferative effects of gliotoxin on human HT1080 fibrosarcoma cells, we treated cultures with various concentrations of gliotoxin (0–100 μM) for 24 h. The antiproliferative effects were assessed by a colorimetric MTT assay. Gliotoxin treatment decreased the number of viable cells in a dose-dependent manner (Fig. 1). This suggested that gliotoxin decreased proliferation of HT1080 cells, and it may have generally cytotoxic effects.
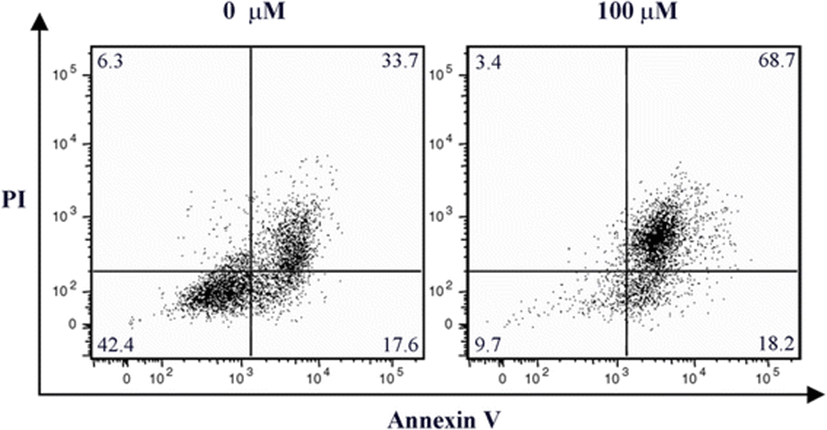
To determine whether the antiproliferative effect of gliotoxin was associated with apoptotic cell death, HT1080 cells were treated with 100 μM gliotoxin for 24 h. Cells were then double stained with Annexin V and propidium iodide (PI), and flow cytometric analyses were performed (Fig. 2). The percentage of Annexin V-positive and PI-positive cells presenting in late-apoptosis phase increased from 33.7 to 68.7 % after treatment with gliotoxin. This clearly indicates that gliotoxin induced apoptosis in HT1080 cells.
NF-κB induces expression of several genes that promote malignant properties associated with tumor growth and inhibit apoptosis (Panwalkar et al. 2004). Constitutive activation of NF-κB has been shown in several types of cancer (Sasaki et al. 2001; Weichert et al. 2007; Sakamoto et al. 2009). In unstimulated cells, NF-κB is typically sequestered in the cytoplasm where it complexes with inhibitors such as IκB-α. When cells experience the proper stimulus, cytoplasmic NF-κB becomes activated primarily through phosphorylation and degradation of IκB-α. NF-κB then translocates from the cytosol to the nucleus and induces expression of target genes (Siebenlist et al. 1994; Baeuerle and Baltimore 1996).
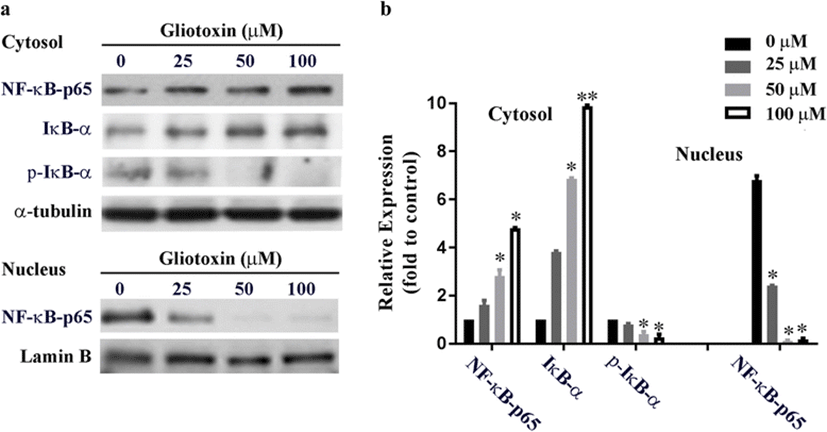
To examine the effects of gliotoxin on NF-κB signaling, we analyzed the expression and phosphorylation state of IκB-α. We also investigated the expression of NF-κB and the nuclear translocation dynamics of the p65 subunit after gliotoxin treatment (Fig. 3). HT1080 cells were cultured with gliotoxin (0–100 μM) for 24 h, and the cytosol and nuclear lysates were each subjected to western blot analysis. Treatment with gliotoxin inhibited NF-κB activation. High levels of NF-κB-p65 were found in nuclear fraction of control HT1080 cells without gliotoxin treatment. However, exposure to gliotoxin decreased the amount of nuclear-localized NF-κB-p65. Gliotoxin treatment also reduced phosphorylation of IκB-α and increased overall expression of IκB-α. Together, these results suggest that gliotoxin inhibited the nuclear import and activation of NF-κB by suppressing the phosphorylation and subsequent degradation of its inhibitor, IκB-α.
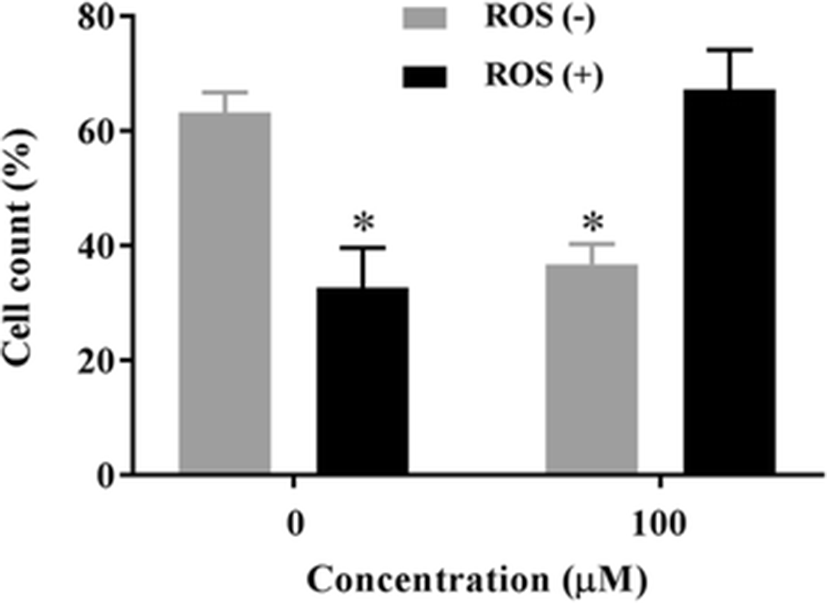
ROS are produced by a variety of cellular processes. This oxidative stress response frequently triggers apoptotic cell death (Russell and cotter 2015). One reason for this is the inhibition of NF-κB activity by ROS, which ultimately enhances apoptosis. Specifically, ROS inhibit DNA binding of NF-κB through direct oxidation of NF-κB (Toledano and Leonard 1991; Nowak et al. 2008) and through the modification of its inhibitor, IκB-α. Therefore, we examined whether gliotoxin influenced ROS levels in HT1080 cells. After treating cells with 100 μM gliotoxin, ROS levels increased from 32.2 to 62.7 % compared to untreated controls (Fig. 4). Taken together, these results suggest that gliotoxin triggered apoptotic cell death in HT1080 cells via generation of ROS and inactivation of NF-κB signaling.
Discussion
Apoptosis is a process of programmed cell death that occurs in multicellular organisms. The development and progression of cancer frequently involves the suppression of apoptosis. Therefore, almost all cytotoxic anticancer therapies currently in clinical use attempt to target cancer cells by inducing apoptosis. The NF-κB pathway is involved in tumorigenesis, tumor cell proliferation, and apoptosis, as well as cellular immunity and inflammation. For example, constitutive activation of NF-κB has been reported in several types of cancers including gastric cancer, pancreatic cancer, and colorectal carcinoma (Sasaki et al. 2001; Weichert et al. 2007; Sakamoto et al. 2009). NF-κB helps activate downstream proteins that promote tumor growth and inhibit apoptosis in tumor cells. Given the antiapoptotic effects of NF-κB, inhibitors of NF-κB are promising antitumor drugs. As a result, much effort has focused on developing inhibitors that target NF-κB (Yemelyanov et al. 2006; Yang et al. 2006). Recently, ROS have been also reported to repress NF-κB signaling in many ways. Direct oxidation of NF-κB by ROS inhibits its DNA-binding ability (Toledano and Leonard 1991). ROS modification of IκB-α leads to inhibition of NF-κB signaling (Nowak et al. 2008). In addition, ROS stabilize IκB-α by inhibiting the proteasome, resulting in downregulation of NF-κB signaling (Wu et al. 2009). Therefore, it seems reasonable that ROS function as physiological signaling modulators of NF-κB signaling cascades in cancers. Our results support this hypothesis. We showed that gliotoxin, a natural compound isolated from the marine fungus A. fumigatus, inhibited proliferation and induced apoptosis in human HT1080 fibrosarcoma cells by increasing intracellular ROS levels and repressing activation and nuclear translocation of NF-κB. Therefore, we suggest that gliotoxin may constitute a promising new antitumor drug for the targeted inhibition of NF-κB signaling.
Conclusions
We demonstrated that gliotoxin, a mycotoxin produced by the marine fungus A. fumigatus, induced apoptosis in HT1080 human fibrosarcoma cells. It achieved this, at least in part, through an increase in levels of intracellular ROS and repression of NF-κB signaling. Cumulatively, our results provide a new avenue of research into the design and development of NF-κB antagonists for the treatment of cancer.