






Introduction
Red sea bream iridoviral disease (RSIVD) was originally identified in Japan in 1990 in red sea bream (Pagrus major) that showed severe anemia with spleen and kidney enlargement (Inouye et al., 1992). The causative agent of RSIVD was later found to be Megalocytivirus of the Family Iridoviridae (Chinchar et al., 2017). According to the World Organisation for Animal Health (OIE), RSIVD is a major cause of mortality in freshwater and marine fish (OIE, 2019). Based on phylogeny of the major capsid protein (MCP) and adenosine triphosphatase (ATPase) genes, Megalocytivirus causing RSIVD is classified into three genotypes, red sea bream iridovirus (RSIV), infectious spleen and kidney necrosis virus (ISKNV), and turbot reddish body iridovirus (TRBIV) (Kurita & Nakajima, 2012). RSIV and TRBIV genotypes have been identified from farmed fishes for RSIVD as endemic in Korea since the 1990s (Do et al., 2005; Jeong et al., 2003; Kim et al., 2019; Oh et al., 2006).
Among several diagnostic assays for RSIVD, bioassays involving virus isolation using cell culture followed by indirect antibody testing or conventional polymerase chain reaction (PCR) amplification, direct PCR analysis, and sequencing have been classified as recommended methods for reasons of availability, utility, and diagnostic specificity (DSp) and sensitivity in the Manual of Diagnostic Tests for Aquatic Animal (OIE, 2019). Real-time PCR assays allow for the detection of amplified signal for PCR amplicons in real-time using fluorescent dyes without the need for agarose-gel electrophoresis and also has advantages of high analyte sensitivity and specificity compared to that of conventional PCR (Arya et al., 2005; Klein, 2002). However, a real-time PCR assay for RSIV as a standard diagnostic assay is yet not provided in the Manual of Diagnostic Tests for Aquatic Animal (OIE, 2019). Several diagnostic assays for megalocytiviruses causing RSIVD based on real-time PCR have been developed and evaluated for their analyte sensitivity and specificity (Kim et al., 2014; Lee et al., 2020; Rimmer et al., 2012). However, their diagnostic sensitivity (DSe) as determined by the proportion of samples from known infected reference animals testing positive in an assay and their DSp as determined by the proportion of samples from known non-infected reference animals testing negative in an assay have not been validated against the conventional PCR assays using field samples.
In the present study, a TaqMan probe-based real-time PCR assay that used a probe and primers designed based on consensus sequences of MCP genes from megalocytiviruses was developed and validated with its DSe and DSp for RSIV being compared against conventional PCR assays. The utility of the new TaqMan probe-based real-time PCR assay for viral quantitative analysis was evaluated through comparison to that of a SYBR-Green real-time PCR assay using RSIV-positive samples.
Materials and Methods
For virus propagation, primary cells derived from rock bream (Oplegnathus fasciatus) were grown in L-15 medium (Gibco, Grand Island, NY, USA) containing 10% Premium Plus fetal bovine serum (FBS; Gibco) and 1% antibiotic-antimycotic solution (Gibco) as described by Lee et al. (2020). The following megalocytiviruses isolated from primary cells were used in the study: RSIV subtype I (17SbTy strain) derived from sea bass (Lateolabrax japonicus), RSIV subtype II (17Rb24Gs strain) derived from rock bream (Kim et al., 2019), ISKNV-type (PGIV strain) derived from pearl gourami (Trichogaster leeri) (Jeong et al., 2008), and TRBIV-type (FLIV strain) derived from olive flounder (Paralichthys olivaceus) (Kim et al., 2014). Viral infections were conducted in 25-cm2 flasks of 80%–90% cell confluency in L-15 medium supplemented with 5% FBS and 1% antibiotic-antimycotic solution at 25°C for 7 days. The supernatants were collected following the appearance of cytopathic effects of rounding cells and centrifuged at 500×g for 10 min and the clarified supernatants stored at −80°C until use.
To design primers and a probe for detecting Megalocytivirus causing RSIVD (RSIV, ISKNV, and TRBIV-types), consensus sequences of 166 MCP genes retrieved from GenBank of the National Center for Biotechnology Information (NCBI) and 39 MCP genes of RSIVs identified from farmed fish in Korea (Kim et al., 2019). The sequences were aligned using BioEdit Ver 7.2.5 software (Fig. 1). Forward primer RSIV 1094F and reverse primer RSIV 1221R were designed according to consensus sequence base positions 1,094−1,112 and 1,199−1,221, respectively (Table 1). The TaqMan probe (RSIV 1177 probe) was designed according to base positions 1,199−1,221 of the consensus sequences and presented a 6°C higher melting temperatures (Tm) than those of the forward and reverse primers. The probe was labeled with carboxyfluorescein (FAM) at the 5′ end as a reporter. Black hole quencher-1 (BHQ-1) was used to label the 3′ end of the probe as a quencher.

| Genomic regions | Primers | Sequence (5'–3') | Amplicon (bp) | Purpose | Reference |
|---|---|---|---|---|---|
| Pst I fragment | 1-F | CTC AAA CAC TCT GGC TCA TC | 570 | Detection1) | Kurita et al. (1998) |
| 1-R | GCA CCA ACA CAT CTC CTA TC | ||||
| DNA polymerase gene | 4-F | CGG GGG CAA TGA CGA CTA CA | 568 | ||
| 4-R | CCG CCT GTG CCT TTT CTG GA | ||||
| Major capsid protein | RSIV-MCP1F | ATG TCT GCR ATC TCA GGT GC | 1,362 | Cloning2) | Kim et al. (2018) |
| RSIV-MCP1362R | TYA CAG GAT AGG GAA GCC TGC | ||||
| MqF | GGC GAC TAC CTC ATT AAT GT | 141 | Detection and quantification3) |
Jin et al. (2014) | |
| MqR | CCA CCA GGT CGT TAA ATG A | ||||
| RSIV 1094F | CCA GCA TGC CTG AGA TGG A | 128 | Detection and quantification4) |
This study | |
| RSIV 1221R | GTC CGA CAC CTT ACA TGA CAG G | ||||
| RSIV 1177 probe | FAM-TAC GGC CGC CTG TCC AAC G-BHQ1 |
Viral nucleic acid of cultured RSIV 17Rb24Gs strain was extracted using a Patho Gene-spinTM DNA/RNA Extraction Kit (Intronbio, Seongnam, Korea) according to the manufacturer’s protocol. PCR amplification of the MCP gene was performed using the primers described in a previous study (Kim et al., 2018) and shown in Table 1. To construct a recombinant plasmid for determination of analyte sensitivity and quantitative analysis as a positive control template, a 1,362 bp PCR amplicon was inserted into pGEM-T easy vector (Promega, Madison, WI, USA) and used to transform Escherichia coli DH5α cells. The plasmid containing the MCP gene was then purified from the transformed E. coli using GeneAll Plasmid SV Mini Kit (GeneAll Biotechnology, Seoul, Korea).
Real-time PCR was performed with a 20 μL final volume reagent mixture containing 1 μL DNA, 200 nM of each primer (RSIV 1094F and RSIV 1221R) and probe (RSIV 1177 probe), 10 μL 2× HS Prime qPCR Premix (Genet Bio, Nonsan, Korea), 0.4 μL 50× ROX dye, and 5.6 μL nuclease-free water using a StepOne Real-time PCR system (Applied Biosystems, Waltham, MA, USA). After pre-denaturation at 95°C for 10 min, amplifications were conducted with 40 cycles of 94°C for 10 s (denaturation) and 60°C for 35 s (annealing and extension).
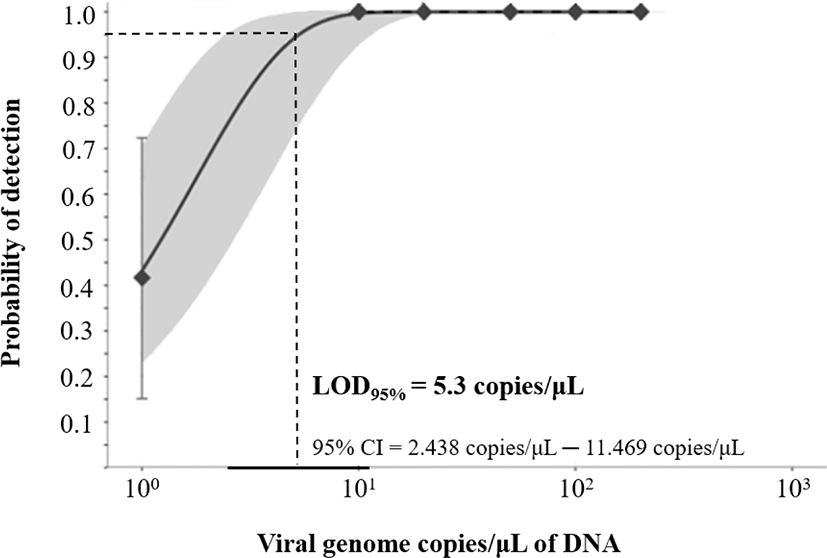
To evaluate analyte specificity of the developed real-time PCR assay, DNA extracted from four different megalocytiviruses (RSIV subtype I, RSIV subtype II, ISKNV, and TRBIV) were used. For RNA viruses hirame rhabdovirus (HIRRV; ATCC VR-1391), viral nervous necrosis virus (VNNV; RGNNV type), and viral hemorrhagic septicemia virus (VHSV; IVa type) described by Kim et al. (2016), complementary DNAs (cDNAs) were synthesized using an UltraScript 2.0 cDNA Synthesis Kit (PCR Biosystems, London, UK) and then used as template for real-time PCR assays. DNA extracted from two bacteria species commonly isolated from marine fish in Korea, Vibrio harveyi isolated from olive flounder and Streptococcus parauberis (FP3287) from olive flounder, was obtained from the Korean Culture collection of Aquatic Microorganisms (KoCAM) of the National Institute of Fisheries Sciences (NIFS) and used as the template for real-time PCR assays. For evaluation of analyte sensitivity, the limit of detection (LOD) was determined using serial-diluted plasmid DNA containing the MCP gene (200, 100, 50, 20, 10, 1 viral genome copies/µL) in twelve replicates using the modified method of Uhlig et al. (2015). LOD95%, the lowest number of viral genome copies at which 95% of samples test positive, was calculated using Quodata software (http://quodata.de). The probability of detection (POD) graph was generated with a 95% confidence interval.
DNAs obtained from 112 farmed fish spleens (10 mg/specimen) and/or kidneys (10 mg/specimen) were collected between 2012 and 2020 and used for analysis in the current experiment. The farmed fish included 58 olive flounder, 27 rock bream, 10 starry flounder (Platichthys stellatus), 8 red sea bream, 7 blackhead sea bream (Acanthopagrus schlegelii), 1 stone flounder (Kareius bicoloratus), and 1 Korean rockfish (Sebastes schlegelii) as detailed in Supplemental Table 1. DNA samples with 260/280 nm absorbance ratios between 1.8 and 2.0 were diluted to 50 ng/µL for use in the experiment. For the qualitative analysis, two OIE-recommended PCR assays (1F/1R and 4F/4R primer sets; Table 1) and the newly developed real-time PCR assay were carried out. The positive control sample was confirmed by positive results for both PCR assays and sequencing analysis. The DSe and DSp were determined as described by Jacobson (1998). For comparison of the quantitative results using the positive control samples, SYBR Green-based real-time PCR was performed using the primers targeting a fragment of the MCP gene (Jin et al., 2014). Briefly, the real-time PCR assay was performed using a reagent mixture containing 1 μL DNA, 200 nM of each primer (MqF and MqR), 10 μL 2× Prime Q-Mastermix (Genet Bio), 0.4 μL 50 × ROX dye, and 6.6 μL nuclease-free water and amplified with a StepOne Real-time PCR System (Applied Biosystems). The cycling profile consisted of 94°C for 10 min followed by 40 cycles of 94°C for 10s, 62°C for 15 s, and 72°C for 20 s. A serial 10-fold dilution of positive control plasmid containing the MCP gene was used to generate a standard curve ranging from 101 to 108 viral genome copies/µL. Statistical significance between the SYBR Green and TaqMan probe-based real-time PCR assays in quantifying the number of viral genome copies was determined according to a paired T-test using GraphPad Prism (Ver. 8.4.3) software. p < 0.05 were considered statistically significant.
Results and Discussion
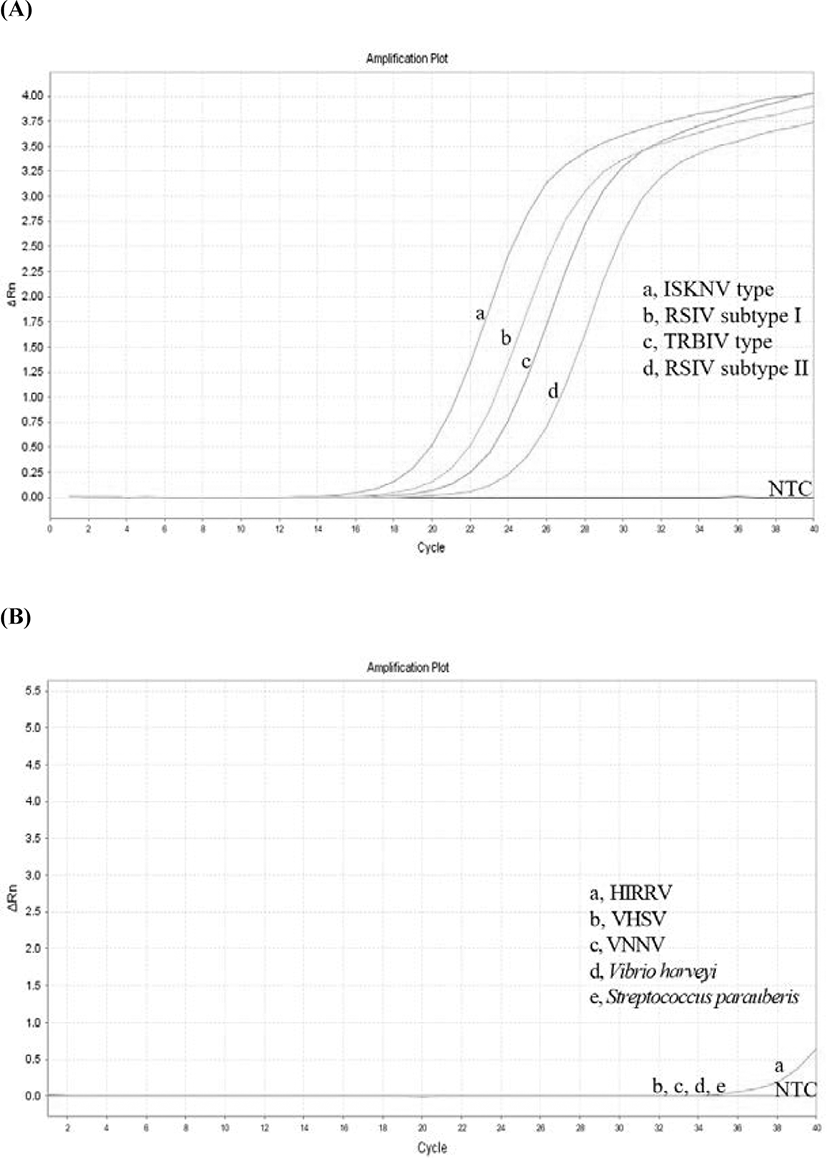
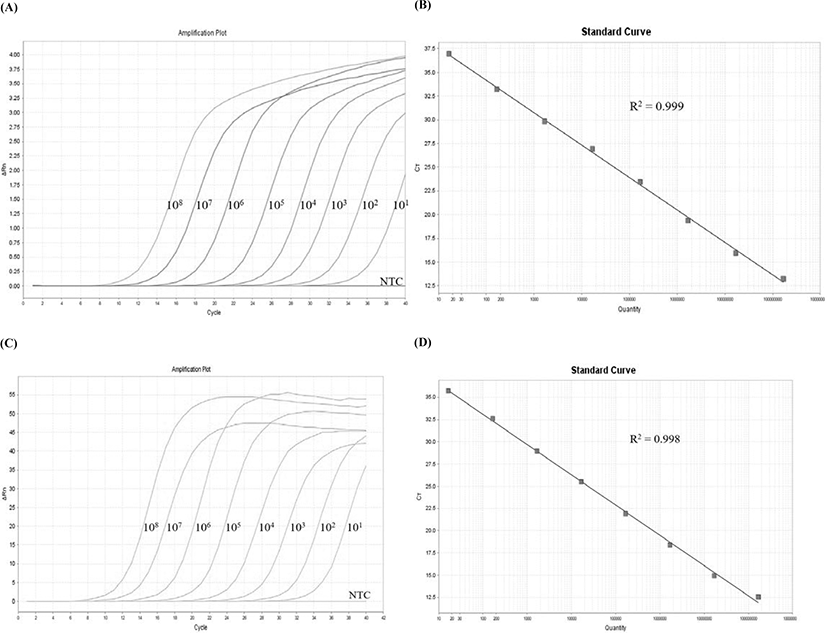
To develop a TaqMan probe-based real-time PCR assay for RSIV, specific primers and probe were designed based on 205 consensus sequences of MCP genes complied from the NCBI database and Korean isolates. The MCP gene is one of the conserved regions that has been used for genotyping based on the phylogeny, as well as used for molecular diagnosis of the genus Megalocytivirus (Jin et al., 2014; Kim et al., 2019; Kurita & Nakajima, 2012; Lee et al., 2020; Song et al., 2008). The amplification plot demonstrated that specific amplicons were generated from RSIV subtype I, RSIV subtype II, ISKNV, and TRBIV-types megalocytiviruses (Fig. 2A). Other pathogens, including VHSV, VNNV, S. parauberis, and V. harveyi, were not amplified using the newly designed primers and probe (Fig. 2B). Fluorescent signals were detected with analysis of HIRRV cDNA after more than 35 cycles of PCR amplification; however, PCR amplicons were not observed with agarose-gel electrophoresis (data not shown). This result suggests the developed real-time PCR assay may be used in specific qualitative analysis for megalocytiviruses including RSIV, ISKNV, and TRBIV. The standard curve results showed high correlation coefficients for both the TaqMan probe PCR (r2 = 0.999; Fig. 3A and 3B) and the for SYBR Green PCR (r2 = 0.998, Fig. 3C and 3D) within the range of 1.68 × 101−108 viral genome copies/μL. This suggested the positive control plasmid DNA could be used for both quantitative analysis and sensitivity analysis with a LOD95% for the lowest number of viral genome copies. In previous studies, detection limits were reported as 1 × 104 viral genome copies/rxn for an OIE recommended PCR assays (Rimmer et al., 2012) and 102–103 viral genome copies/uL for a peptide nucleic acid (PNA)-based real-time PCR assay (Lee et al., 2020). Although comprehensive validations using artificially infected samples by infection stages are needed, the detection limit of the developed real-time PCR assay was 0.5 pg/μL of DNA (corresponding to 2.5 viral genome copies/μL) isolated from an RSIV-infected rock bream in the field (data not shown). Furthermore, the LOD95% was determined to be 5.3 viral genome copies/μL of positive plasmid DNA containing the MCP gene from RSIV (95% confidence interval, 2.438–11.469 viral genome copies/μL; Fig. 4), suggesting this assay could be applied with samples possessing low viral levels (approximately 10 viral genome copies), such as environmental water samples, reservoir samples, and biopsy samples collected during the initial stages of infection.
The proportion of true positive and true negative samples evaluated by a diagnostic assay is identified according to DSe and DSp, respectively. Furthermore, DSe and DSp can be applied as indicators for verification of a diagnostic assay to estimate the status of pathogen infection or exposure in an individual or group (Jacobson, 1998; Saah & Hoover, 1997). Detection results for a total of 112 samples using conventional PCR and the developed real-time PCR assays were in agreement in which 28 samples being true positives and 80 samples being true negatives (Table 2). Only 4 samples presented different results between the assays. Based on the formulas [DSe = true positive (TP) / TP + false negative (FN); DSp = true negative (TN) / TN + false positive (FP)] described by Jacobson (1998), DSe and DSp of the developed real-time PCR assay were 100% and 95.2%, respectively. This result indicated that the developed real-time PCR assay can be efficiently utilized for RSIV detection in parallel with conventional PCR assay.
| Comparison of the assays | Positive1) | Negative1) | TaqMan probe real-time PCR assay |
|---|---|---|---|
| Positive2) | 28 | 4 | 32 |
| Negative2) | 0 | 80 | 80 |
| PCR assays | 28 | 84 | 112 |
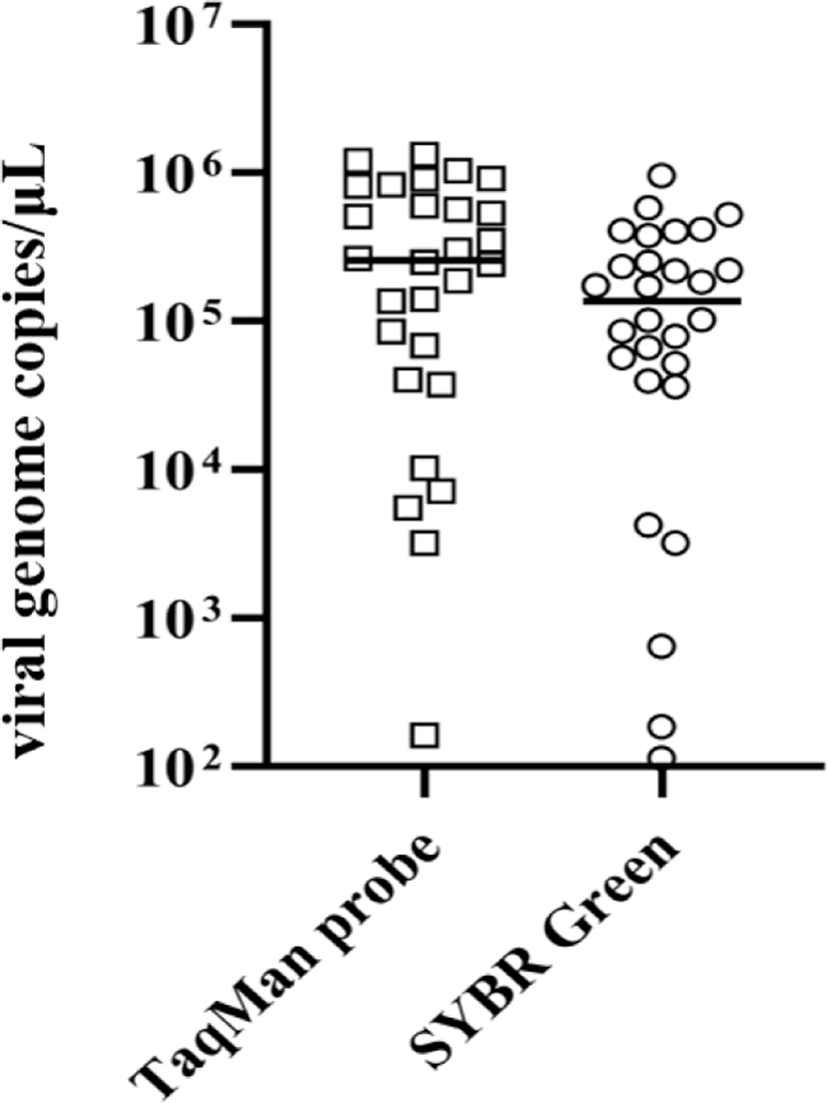
Among fluorescent dyes generating amplification signals from PCR amplicons in real-time PCR assays, SYBR Green and the TaqMan probe are generally used for quantitative analysis (Arya et al., 2005). SYBR Green dye, which binds double-stranded DNA by intercalating between the DNA bases during the extension step of PCR amplification, has several advantages over TaqMan probe, including easier primer design and less expensive operation cost. However, SYBR Green-based assays may produce false-negative results caused by non-specific amplicons or primer-dimerization (Arikawa et al., 2008). TaqMan probes are oligonucleotide consisting of reporter and quencher dyes and demonstrate greater DNA binding affinity than that of SYBR Green (Wilhelm & Pingoud, 2003). Thus, the TaqMan probe-based real-time PCR assay would be expected to be a more appropriate method of qualitative and quantitative virus analysis in terms of specificity and sensitivity. Results of the number of viral gene copies in the 28 RSIV-positive samples was not significantly different (p > 0.05) between the SYBR Green and TaqMan probe-based real-time PCR assays (Fig. 5, Supplemental Table 2). This indicated the newly developed real-time PCR assay was sufficient for use in quantitative analyses.
In conclusion, we developed a specific and highly sensitive (LOD95%, 5.3 viral gene copies/μL) TaqMan probe-based real-time PCR assay for RSIV based on primers and probe designed using consensus sequences of MCP genes. Based on the comparative analysis to that of conventional PCR assays, the DSe and DSp of the developed real-time PCR assay for RSIV were 100% and 95.2%, respectively. Furthermore, the determination of the number of viral genome copies in RSIV-positive samples did not significantly differ between the SYBR Green and TaqMan probe-based real-time PCR assays. These results suggest that our developed real-time PCR assay may be used as a robust diagnostic tool for qualitative and quantitative analysis of RSIV.