






Introduction
The decline in biodiversity in freshwater ecosystems is greater than that in many other ecosystems because of rising human demand for certain of the organisms therein over the past century. This biodiversity crisis requires priority setting at global, regional, and local scales to concentrate limited resources to address the most pressing conservation issues. Endemic species, which are conservation priorities, are typically seen in freshwater habitats due to cause evolution of many species with small geographic range (Strayer & Dudgeon, 2010). Therefore, endemic species are primary candidates for extinction (Anderson, 1994). Especially, molluscs have higher extinction rates than any other taxa, making them the most threatened group of animals on earth (Régnier et al., 2009). Efficient resource management requires management of population structure based on genetic research into species and populations (Kang et al., 2010). To this end, the Ministry of Environment of the Republic of Korea (ROK) has identified Korean peninsula-endemic organisms and operates an approval system that must be satisfied before they can leave the country. Among the taxa endemic to the Korean peninsula, the genus Semisulcospira is a speciose family of freshwater gastropods distributed throughout eastern Asia, including in the ROK, Japan, China, and Taiwan (Chiu et al., 2017). Although five species of the genus Semisulcospira inhabit the ROK, only three (Semisulcospira coreana, Semisulcospira forticosta and Semisulcospira tegulata) except Semisulcospira gottschei, have been designated as endemic species by the Ministry of Environment. Semisulcospira is a major shellfish for domestic inland fisheries, as evidenced by an annual output of 574 tons and annual production value of 6.4 billion Korean won, second only to the family Viviparidae (http://kostat.go.kr). Moreover, the snails are intermediate hosts of human pathogens and are used as a biological marker of the environmental status of freshwater systems (Pan et al., 2011). Despite their important role, there were still the insufficient taxonomic system and genetic population structure. Until now, the morphology, reproductive anatomy, and karyotypic, electrophoretic, and mitochondrial variation of this snail species have been analyzed but its genetic population structure remains unclear (Jung et al., 1999; Kim et al., 2012). Therefore, investigation of within-species genetic diversity is needed to obtain information on population migration and history.
Studies of genetic population structure have sought to improve the management and preservation of freshwater resources. To date, most genetic diversity studies have used molecular markers include allozyme, mitochondrial DNA (mtDNA), also developments in molecular-biological technology have enabled large amounts of genetic information to be processed (Park et al., 2021). This genetic information facilitates the development of new markers for analysis of populations of marine organisms. In analyses of genetic diversity, the sensitivity of the marker gene determines its performance, and mtDNA is typically used due to its high sequence variation (Dunham, 2004). This variation enables the exploration of parameters such as subdivision and gene flow (Hillis et al., 1996). The mitochondrial cytochrome c oxidase subunit I (COI) gene (1,500 bp) has been used as a DNA barcode for population studies of fish and shellfish because of its marked nucleotide variation (Ward et al., 2005). In addition, the NADH dehydrogenase subunit 4 (ND4) gene has been used as a marker for analyzing the genetic population structure of fish and shellfish (Doosey et al., 2010). The population genetic structure of shellfish using the mtDNA COI gene has been variously studied (Arnaud et al., 2000; Matsumoto, 2003). However, our understanding of intraspecies genetic variation by geographic area for this snail using the only one mtDNA maker is limited.
In this study, we analyze the genetic characteristic such as genetic diversity among four Semisulcospira species endemic to Korea. This is the first study of population genetic structure of four Semisulcospira species in Korea, and this study will contribute useful genetic information for the management of geographically separated populations of this snail.
Materials and Methods
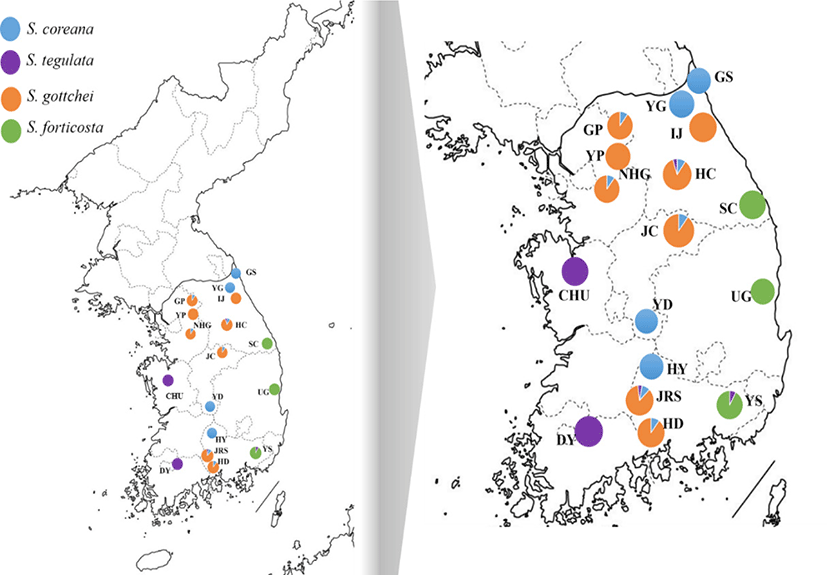
The genus Semisulcospira were obtained from 19 localities in the ROK from December 2015 to August 2019. Information on the samples is shown in Fig. 1. We collected muscle tissue samples from stored by the National Institute of Fisheries Science (NIFS) or fish markets. The samples were preserved in 99% ethanol until use. Genomic DNA was extracted using the DNeasy Blood and Tissue® Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Briefly, ~20 mg of tissue was lysed in Buffer ATL containing proteinase K (Qiagen) at 56℃ for 3 h. The lysates were mixed with Buffer TL and ethanol, and DNA was purified using a resin column. The concentration of genomic DNA was estimated using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Amplification and sequencing of a partial region of the COI gene were performed using the universal primers HCO2198 (5’-TAA ACT TCA GGG TGA CCA AAA AAT CA-3’) and LCO1 490 (5’-GGT CAA CAA ATC ATA AAG ATA TTG G-3’).
Polymerase chain reaction (PCR) amplification of the COI and ND4 genes was performed as follows: initial denaturation at 94℃ for 7 min, followed by 35 cycles of denaturation at 94℃ for 45 s, annealing at 56℃ and 60℃ for 45 s, extension at 72℃ for 45 s, and a final extension at 72℃ for 7 min. PCR was performed on a total solution of 10 μL containing 1 μL of DNA, 0.25 U of Ex Taq DNA polymerase, 10 × PCR buffer, 0.2 mM dNTP, and 10 pmol of each primer. The PCR products were purified using the DavinchTM PCR Purification Kit (Davinch-K, Seoul, Korea). Finally, the PCR products were sequenced using an ABI 3730XL sequencer (Applied Biosystems, Foster City, CA, USA) and an ABI Prism BigDyeTM Terminator Cycle Sequencing Ready Reaction Kit v. 3.1 (Applied Biosystems).
To identify within- and between-species mutations and preserved regions in the ND4 gene, the mtDNA sequences of S. coreana (NC037771.1 and LC333861.1) and Semisulcospira libertina (NC023364.1 and KF736848.1) in NCBI GenBank were aligned using BioEdit software (http://mbio.ncsu.edu/BioEdit/bioedit.html). Based on the conserved region, specific forward and reverse primers were developed to amplify the ND4 gene. The information of species-specific ND4 primers was and SCSL-ND4-180F (5’-TAC CCT TAT YTC YYT AAC TTT TTG-3’) and SCSL-ND4-739R (5’-AAA ATT GAT GTG AAC GWA GGA-3’).
The acquired sequences were assembled using ClustalW in SeqMan software (DNASTAR, Madison, WI, USA). A maximum likelihood tree using TPM3u + F + I + G4 model with bootstrap analysis was constructed using IO-Tree software (Nguyen et al., 2015). The resulting Newick-formatted file were visualized in iTOL v 5.2 (http://itol.embl.de/). Haplotype diversity (h) and nucleotide diversity (π) were estimated using ARLEQUIN v. 3.5.1.2 (http://lgb.unige.ch/arlequin/software/). Demographic history was estimated based on the Tajima’s D and Fu’s Fs values, obtained by neutrality testing in ARLEQUIN v. 3.5.5. The Bayesian skyline plot (BSP) method implemented in BEAST2 (Bouckaert et al., 2014; Drummond et al., 2005). The general time-reversible model of nucleotide substitution was selected for all four species. Coalescent genealogies of BSP were constructed with a strict clock model for a total of 100 million generations, sampling every 1,000 steps. Convergence to the stationary distribution and sufficient effective population sizes for each estimated parameter were checked using Tracer v.1.7.1 (Rambaut et al., 2018).
Analysis of molecular variance (AMOVA) was performed to assess differentiation between populations using ARLEQUIN v. 3.5.1.2. The significance of pairwise FST values was calculated at the 0.05 level based on 10,000 permutations. A minimum spanning network was generated from the COI and ND4 genes (942 bp) using PopArt v. 1.7 software (http://popart.otago.ac.nz), with epsilon set to zero.
Results
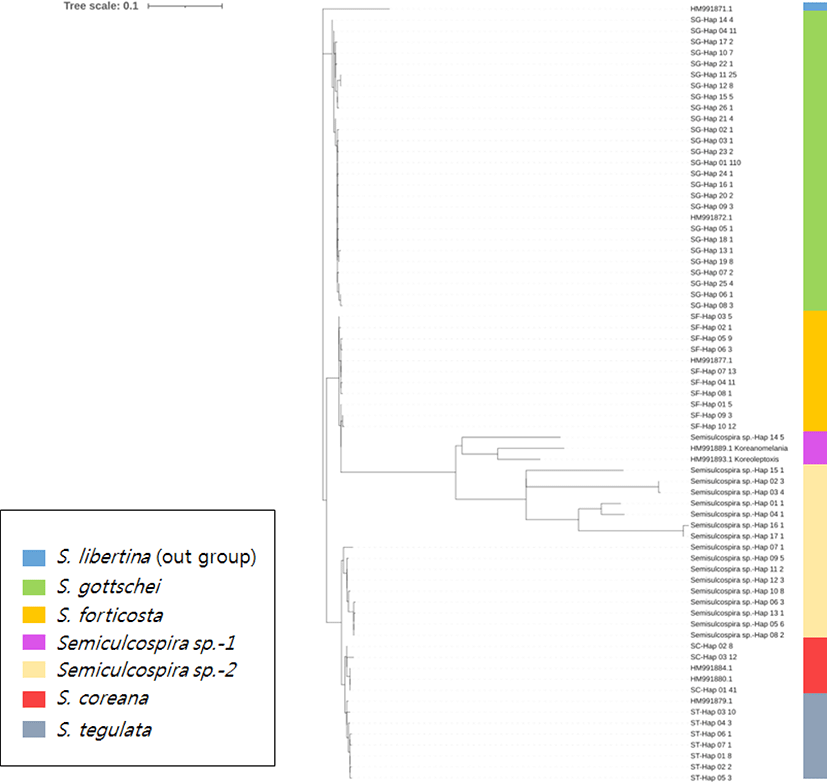
The collected each samples were identified by the sequence analysis of the mtDNA COI gene by using the references stored in the Storage of National Fisheries Research and Development Institute through the study by Kim et al. (2010) and has been submitted to GeneBank (Acession No. HM991871– HM991887). This reference samples identified based on their shell morphology and genetic analysis. Samples identified as Semisulcospira sp. that were genetically different from each clade were excluded in further analysis. Phylogenetic tree indicates genetic distance among all sample of genus Semisulcospira (Fig. 2).
A total of 61 S. coreana individuals of 10 populations was obtained from the 456 bp mtDNA COI gene fragment. Although the target size of COI fragment was 658 bp, readable sequences from some samples were shorter and we used fragment of 456 bp. Three haplotypes were detected only among COI genes; haplotype 1 (Hap_1) predominated (67.2%). Eight haplotypes were detected in the ND4 gene (486 bp) from 58 samples of S. coreana, with Hap_1 being predominant (70.4%). Analysis of the COI + ND4 (942 bp) identified 11 haplotypes among 58 individuals from 10 locations. We could not get the sequence analysis result 3 out of total 61 samples and the rest 58 samples were used for further analysis. COI + ND4 yielded a higher haplotype diversity than COI and ND4 in each population (Table 1). The Gapyoung, Gangwon-do (GP), Mt. Jiri (JRS), and Hongcheon, Gangwon-do (HC) populations displayed only one haplotype for all of the genetic markers (COI, ND4, COI + ND4). The Yeoungdong, Jeollanam-do (YD) population had a higher level of haplotype diversity than the other populations for ND4, COI + ND4 genetic markers. The nucleotide diversity of the YD population was higher than that of the other populations for ND4, COI + ND4 genetic markers.
n, number of specimens; N, number of haplotypes; h, haplotype diversity; π, nucleotide diversity; mtDNA, mitochondrial DNA; COI, cytochrome c oxidase subunit I; ND4, NADH dehydrogenase subunit 4; YG, Yanggu, Gangwon-do; GS, Goseong, Gangwon-do; YD, Yeoungdong, Jeollanam-do; HY, Hamyang, Gyeongsangnam-do; GP, Gapyoung, Gangwon-do; JC, Jaecheon, Chungcheongbuk-do; JRS, Mt. Jiri; HD, Hadong, Gyeongsangnam-do; NHG, Namhan river; HC, Hongcheon, Gangwon-do.
The results of neutrality tests are shown in Table 1. Fu’s Fs value was negative for all populations (excluding those with only one haplotype) and those of the Jaecheon, Chungcheongbuk-do (JC) and Namhan river (NHG) populations were not significant (p > 0.05). Also, Tajima’s D test yielded a negative result for the Yanggu, Gangwon-do (YG), Goseong, Gangwon-do (GS), and Hadong, Gyeongsangnam-do (HD) populations (p > 0.05).
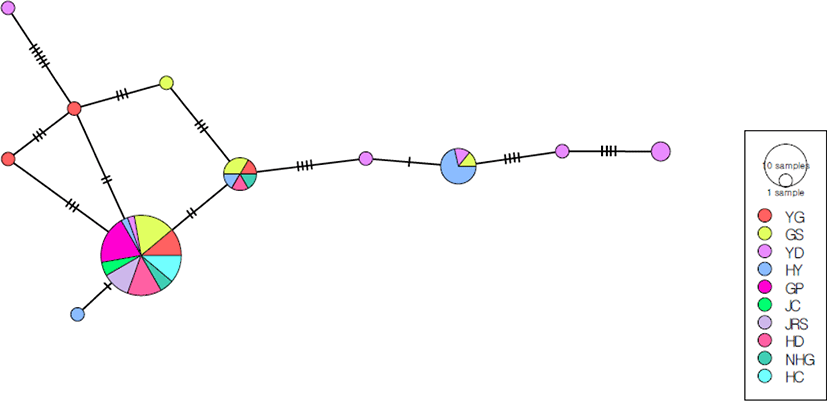
The phylogeny of the 11 haplotypes of the COI + ND4 sequence with no visible geographic population structure was assessed using the minimum spanning algorithm (Fig. 3). The number of haplotypes per population ranged from one to seven (Table 1). The predominant haplotype was present in all 10 S. coreana colonies (62% of all individuals). The second most common haplotype was present in 12% of all individuals, but was shared only by the Hamyang, Gyeongsangnam-do (HY), YD, and GS populations. The YD, YG, HY, and GS populations had 4, 2, 1, and 1 unique haplotype, respectively.
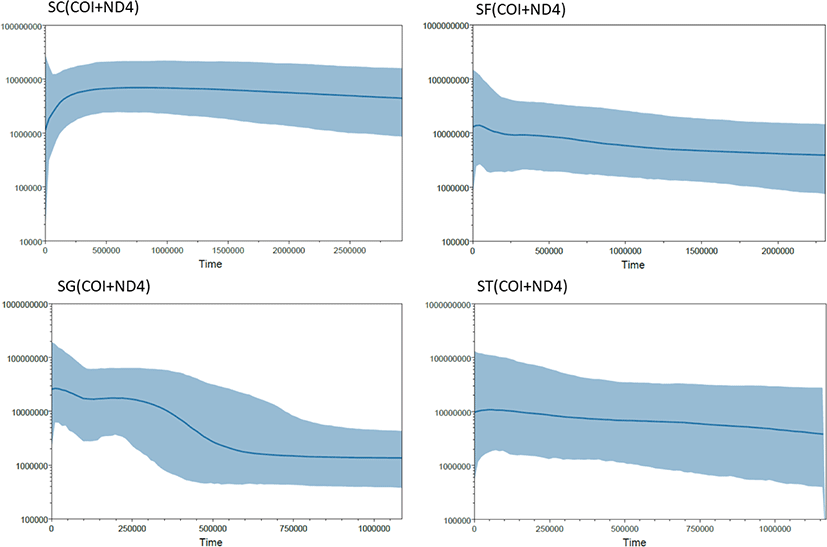
The BSPs showed that the population size decreased slightly for S. coreana (Fig. 4). The estimated mutation rate of Semisulcospira species were 1.06 × 10–9 substitution per year, which is comparable to the mutation rate in other organisms in diverse taxonomic group (Lynch, 2010).
AMOVA showed that the variation among populations (36.1%) was lower than that within populations (63.9). The overall FST value (0.36098) indicated weak genetic differentiation among 10 populations (Table 2). The pairwise FST values of the COI + ND4 sequence ranged from −0.007 to 0.620 (Table 3). The HY and YD populations exhibited significantly greater pairwise genetic differentiation than the other populations. The YG and GS populations had the lowest genetic divergence, and the HY and GP populations the highest.
| Source of variation | df | Sum of squares | Variance components | Percentage of variation (%) |
|---|---|---|---|---|
| Among populations | 9 | 44.282 | 0.65922 | 36.1 |
| Within populations | 48 | 56.051 | 1.16698 | 63.9 |
| Fixation | Index | F ST | 0.36098 |
The total number of specimens analyzed was 63 for COI, 53 for ND4, and 53 for COI + ND4. In total, 12, 7, and 25 mtDNA haplotypes were obtained from the COI, ND4, and COI + ND4 sequences for the three S. forticosta populations, respectively (Table 4). Haplotype diversity was typically high, ranging from 0.529 to 0.949. The Yangsan, Gyeongsangnam-do (YS) population had higher haplotype diversity and nucleotide diversity than the other populations based on the COI + ND4 sequence (h = 0.949; π = 0.00301). The Samcheok, Gangwon-do (SC) population had the second-highest haplotype diversity based on all genetic markers (0.636–0.927).
Neutrality testing was performed to determine the variation in population sizes. Tajima’s D test based on the ND4 gene yielded a positive value for all populations except Ulgin, Gyeongsangbuk-do (UG) (p > 0.05). However, Fu’s Fs values differed significantly among the markers (p < 0.05). Additionally, Fu’s Fs values were negative for all markers in all populations, indicating expansion. The BSPs showed that the population size increased for S. forticosta (Fig. 4).
A minimum-spanning parsimony analysis was performed based on the COI + ND4 mtDNA haplotypes of three S. forticosta populations (Fig. 5). There was no dominant haplotype in which the network contained all populations, and haplotypes with only two populations were observed. Also, the networks were clustered into three distinct groups: SC, YS, and UG. Most of the UG groups were branching out based on the haplotype of SC.
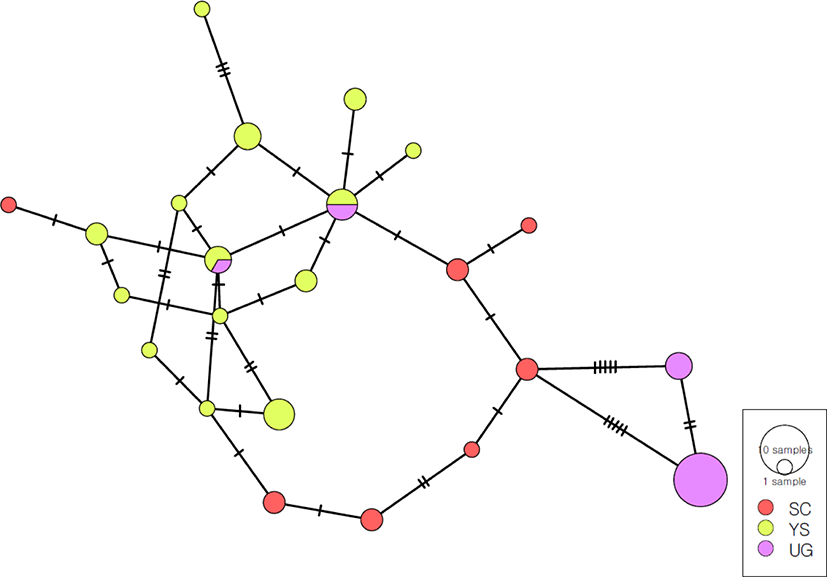
AMOVA showed that the variation among populations (54.82%) was higher than that within populations (45.18%). The overall FST value (0.54815) indicated genetic differentiation among SC, YS, and UG (Table 5). Pairwise comparison revealed a high degree of genetic structure among the populations (Table 6). The three pairwise comparisons yielded significant FST values (0.314–0.624). The YS and UG populations had the highest genetic differentiation, and the YS and SC populations the lowest.
| Source of variation | df | Sum of squares | Variance components | Percentage of variation (%) |
|---|---|---|---|---|
| Among populations | 2 | 58.168 | 1.6439 | 54.82 |
| Within populations | 50 | 67.754 | 1.35508 | 45.18 |
| Fixation | Index | F ST | 0.54815 |
| SC | YS | UG | |
|---|---|---|---|
| SC | 0.000 | ||
| YS | 0.314* | 0.000 | |
| UG | 0.550* | 0.624* | 0.000 |
n, number of specimens; N, number of haplotypes; h, haplotype diversity; π, nucleotide diversity; mtDNA, mitochondrial DNA; COI, cytochrome c oxidase subunit I; ND4, NADH dehydrogenase subunit 4; IJ, Inje, Gangwon-do; YP, Yangpyoung, Gangwon-do; GP, Gapyoung, Gangwon-do; JC, Jaecheon, Chungcheongbuk-do; JRS, Mt. Jiri; HD, Hadong, Gyeongsangnam-do; NHG, Namhan river; HC, Hongcheon, Gangwon-do.
The S. gottschei specimens consisted of eight populations (Inje, Gangwon-do [IJ], Yangpyoung, Gangwon-do [YP], GP, JC, JRS, HD, NHG, and HC) were 208 of COI, 183 of ND4, and 183 of COI + ND4. In total, 26, 21, and 40 mtDNA haplotypes were obtained from the COI, ND4, and COI + ND4 sequences, respectively, for the eight populations of S. gottschei. The number of haplotypes in each group was highest for COI, with 11 in the GP population, followed by 10 in JRS and HC, 7 in HD, 5 in NHG and YP, followed by in JC and IJ. There were nine ND4 haplotypes in the HD population, eight in JRS and HC, five in YP, four in GP, and in JC and NHG; IJ contained only one haplotype. Finally, the number of COI + ND4 haplotypes was in the HC population, 12 in JRS and HD, 9 in YP, 7 in GP, 6 in NHG, 4 in JC, and 3 in IJ (Table 7). The haplotype diversity and nucleotide diversity of COI were higher in the GP population than in the other populations (h = 0.868; π = 0.0054). For ND4 and COI + ND4, the HC population had the highest haplotype diversity and nucleotide diversity (h = 0.775; π = 0.00275 and h = 0.876; π = 0.00374, respectively).
Fu’s Fs and Tajima’s D test were performed to assess demographic history (Table 7). Tajima’s D values were negative for all populations based on all genetic markers, except for the HC population based on COI. However, based on all genetic markers, most populations differed from the extreme expansion model (p > 0.05), with the exceptions of IJ based on COI; JRS, HD, and NHG based on ND4; and JC, JRS, and NHG based on COI + ND4. Fu’s Fs test yielded negative or zero values. The BSPs showed that the population size extremely increased for S. gottchei (Fig. 4).
The minimum spanning network was star-like, with a central haplotype and sporadic haplotypes connected by several mutational steps; the 40 haplotypes were separated into two major clusters (Fig. 6). The first cluster consisted of specimens from all eight populations, including a central haplotype and unique haplotypes derived from the main haplotype. The predominant haplotype was present in all eight penguin colonies (48.6% of all individuals). The central haplotype was present at the highest frequency in the JC population, followed by NHG, JRS, HD, HC, and YP (1.12%). The second cluster consists of individuals from six (HC, HD, JRS, GP, YP, JC) of all population. The second cluster consisted of individuals from the HC, HD, JRS, GP, YP, and JC populations. The second most common haplotype was present in 7% of all individuals, and was shared among the HC, HD, JRS, and GP populations.
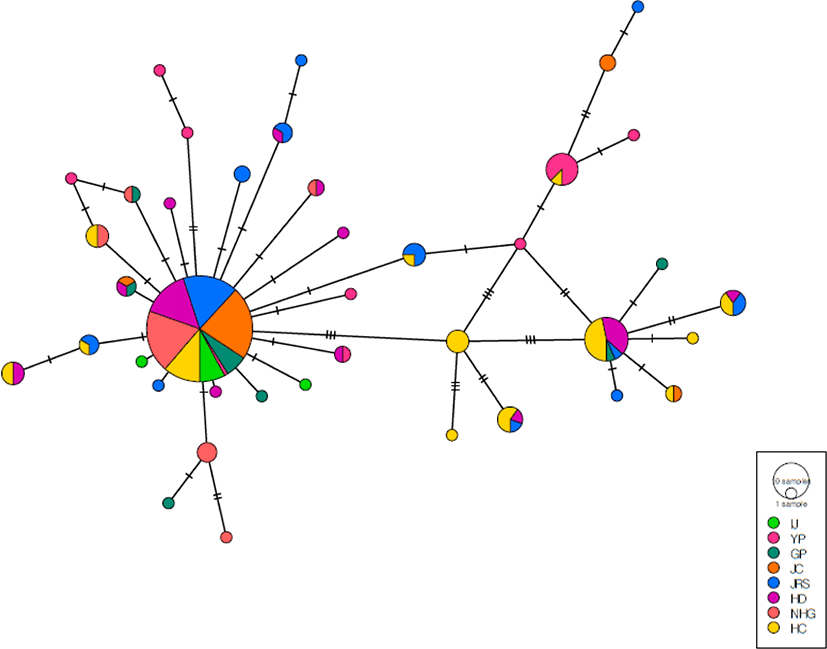
In addition, the network showed many unique haplotypes in each AMOVA showed that the variation among populations (11.73%) was lower than that within populations (88.27%) (Table 8). The overall FST value (0.117) showed that genetic differentiation among the eight populations was low.
| Source of variation | df | Sum of squares | Variance components | Percentage of variation (%) |
|---|---|---|---|---|
| Among populations | 7 | 31.319 | 0.1498 | 11.73 |
| Within populations | 175 | 197.348 | 1.1277 | 88.27 |
| Fixation | Index | F ST | 0.117 |
The pairwise FST values ranged from −0.024 to 0.397 (Table 9). The YP population showed significant differences from the other populations, while the HC population showed significant differences from the other populations with the exception of the HD population.
The 23 S. tegulata individuals in the Damyang, Jeollanam-do (DY), Chungcheongbuk-do (CHU), JRS, HC, and YS populations were analyzed, and the CHU population was found to have the largest number of specimens (Table 10). In total, 7, 11, and 15 mtDNA haplotypes were obtained from the COI, ND4, and COI + ND4 sequences, respectively, in the eight S. forticosta populations. The DY population had three haplotypes based on three genetic markers and the CHU population encompassed 6, 7, and 10 haplotypes based on the COI, ND4, and COI + ND4 sequences, respectively. The JRS population exhibited one, two and one haplotype based on the COI, ND4, and COI + ND4 sequences, respectively. In addition, the HC and YS populations displayed only one haplotype based on the COI, ND4, and COI + ND4 sequences. Haplotype diversity was high, except in populations with only one haplotype. The haplotype diversity of all genetic markers was higher in the CHU population than in the other populations (h = 0.846, 0.872, and 0.961, respectively), but nucleotide diversity was greatest in the DY population (π = 0.00548, 0.0048, and 0.00513, respectively).
n, number of specimens; N, number of haplotypes; h, haplotype diversity; π, nucleotide diversity; mtDNA, mitochondrial DNA; COI, cytochrome c oxidase subunit I; ND4, NADH dehydrogenase subunit 4; DY, Damyang, Jeollanam-do; CHU, Chungcheongbuk-do; JRS, Mt. Jiri; HC, Hongcheon, Gangwon-do; YS, Yangsan, Gyeongsangnam-do.
Fu’s Fs and Tajima’s D test were conducted to evaluate differences in population size (Table 10). The Tajima’s D of the CHU population based on all genetic markers were non-significantly negative. The Fu’s Fs values of the DY and CHU populations based on all genetic markers were negative, but the value of the DY population based on the COI + ND4 sequence was not significant. The BSPs showed that the population size continuously increased and stable for S. tegulata (Fig. 4).
The minimum spanning network of 15 haplotypes based on the COI + ND4 sequence consisted of many unique haplotypes (Fig. 7). The predominant haplotype includes the DY, JRS, HC populations, and appears to be genetically distant from the other haplotypes. The CHU population had 10 unique haplotypes, and there were also haplotype structures that were unique to the DY group. The YS population had one haplotype that was not shared with any other population. The haplotype network suggested that the JRS and HC populations were genetically distant from the other local populations.
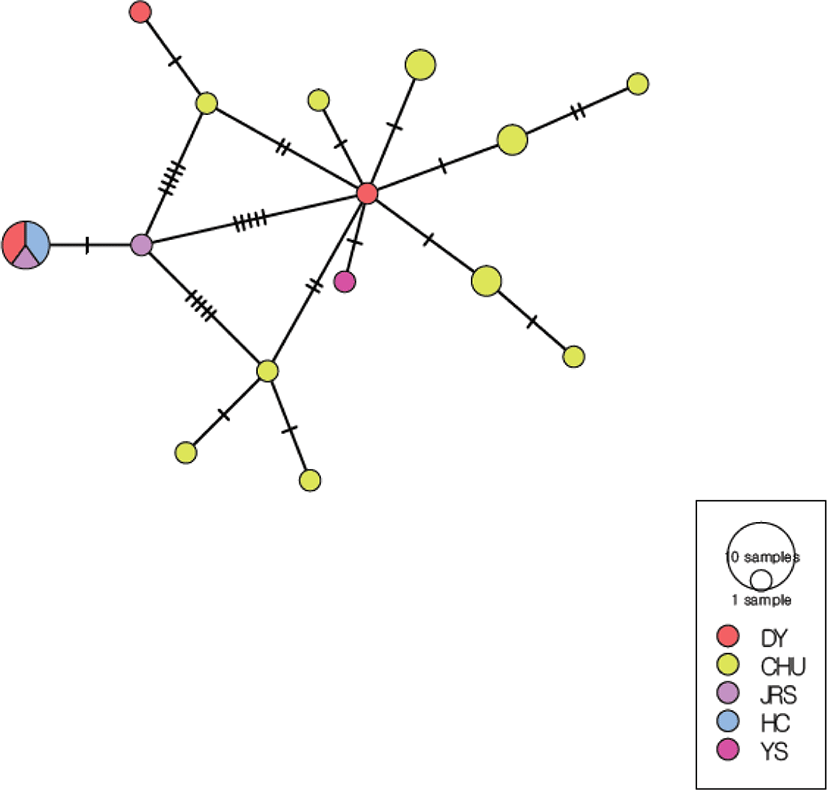
AMOVA showed that the variation among populations (43.58%) was lower than that within populations (56.42%). The overall FST value (0.436) indicated marked genetic differentiation among five populations (Table 11). The pairwise FST values ranged from −0.074 to 0.913. Importantly, the pairwise FST values between geographic localities were not significant for all populations, with the exceptions of the CHU and DY, CHU and JRS, and CHU and HC populations (Table 12).
| Source of variation | df | Sum of squares | Variance components | Percentage of variation |
|---|---|---|---|---|
| Among populations | 4 | 22.038 | 1.124 | 43.58 |
| Within populations | 18 | 26.188 | 1.455 | 56.42 |
| Fixation | Index | F ST | 0.436 |
| DY | CHU | JRS | HC | YS | |
|---|---|---|---|---|---|
| DY | 0.000 | ||||
| CHU | 0.274* | 0.000 | |||
| JRS | –0.074 | 0.587* | 0.000 | ||
| HC | 0.021 | 0.634* | 0.000 | 0.000 | |
| YS | 0.303 | 0.143 | 0.824 | 0.913 | 0.000 |
Discussion
We estimated the level of genetic diversity and historical gene flow among populations of four Semisulcospira species. In the previous study reported that Japanese Semisulcospira species are essentially undifferentiated in the nuclear 28S gene (Köhler, 2016). In this study, the COI, ND4 and COI + ND4 sequences showed that the genetic characterizations of the four species varies throughout the ROK. Nucleotide and haplotype diversity provide demographic information on the history of populations (Grant & Bowen, 1998). A high level of haplotype diversity and low level of nucleotide diversity were observed in 18 populations, with the exception of the S. coreana YD population (Tables 1, 4, 7, and 10). Similar patterns have been reported for other freshwater snail species, such as Lunella granulate (Chiu et al., 2013) and Succinea caduca (Holland & Cowie, 2007). A high level of haplotype diversity is associated with high gene flow among populations (Katsares et al., 2008). In contrast, low nucleotide diversity values indicate small differences between haplotypes (Khosravi et al., 2019), possibly due to expansion after a period characterized by a low effective population size (De Jong et al., 2011). The intraspecific variation of the four snail species in this study was very low (0.05%–0.08%), suggesting small differences among haplotypes. In addition, negative Fu’s Fs and Tajima’s D values imply excess low-frequency polymorphisms, possibly due to demographic expansion. Fu’s Fs and Tajima’s D tests were performed to assess the demographic history of the populations. Negative values indicate excess rare haplotypes and nucleotide site variants under a neutral model of evolution, whereas positive values indicate a recent population bottleneck (Puvanasundram et al., 2018). The four species showed negative Fu’s Fs and Tajima’s D values, except for populations with one haplotype, especially result of Fu’s Fs with a significant (Tables 1, 4, 7, and 10). Fu’s Fs value is regarded as more powerful than Tajima’s D value for detecting the signatures of past population expansions (Fu, 1997). Population expansion is supported by a unimodal pattern of negative Fu’s Fs and Tajima’s D values, together with a high level of haplotype diversity and low level of nucleotide diversity (Chen et al., 2004). Despite the concern over resource reduction in the ROK due to overharvesting, habitat destruction, and human consumption of the four Semisulcospira species, all four species showed population expansion.
The BSP analysis showed that the population expansion generally for the three Semisulcospira species, expect S. coreana (Fig. 4). In S. coreana, the population size decreased relatively recently. This result is thought to have been influenced by the insufficient number of samples from S. coreana populations than in other species populations. Also, it is thought that S. coreana need to manage resources compared to other species. The BSP for other three Semisulcospira species suggest nearly constant or expansion of population size (Fig. 4).
In this study, minimum spanning networks based on the COI + ND4 sequence were constructed to investigate the intraspecific relationships of the four Semisulcospira species. The size of the circle is proportional to the number of specimens with that haplotype, and a single line connecting circles represents a one-base difference. The S. coreana and S. gottschei network consisted of the central haplotype included all populations and several unique haplotypes (Figs. 2 and 5). In the S. coreana network, each haplotype was located around the central haplotype, indicating a greater genetic distance between haplotypes compared to the other three species. The YD population had the lowest frequency (2.7%) of the main haplotype, while the rare haplotype had the most with four, compared to the other S. coreana populations. This is agreement with the corresponding pairwise FST values (Table 3). The S. gottschei network was separated into 40 haplotypes, a larger number compared to the other three species. Also, the S. gottschei network formed two clusters, but it also showed many unique haplotypes. The frequency of the main haplotype in the YP population was 1.12% (Fig. 6). The S. forticosta network showed that this species was the only one with clear regional genetic differences, and no obvious central haplotype. In addition, multiple unique haplotypes were detected at each location, and the UG population was more genetically distinct than the SC and YS populations (Fig. 5). For S. tegulata, no haplotype was common to all populations, and there were only three penguin colonies, which were genetically distant from the CHU population (Fig. 7). Therefore, all populations of the four Semisulcospira species had a large number of unique haplotypes, indicating an excess of rare haplotypes and suggesting rapid genetic differentiation and population expansion. The minimum spanning network did not reveal any haplotypes restricted to specific geographic regions, except for S. forticosta, suggesting a weak phylogeographic structure. This is in accordance with the results of studies on other snail taxa (Nehemia et al., 2019).
The AMOVA, pairwise and overall FST values, based on the COI + ND4 sequences, indicated genetic differentiation of the Semisulcospira populations. AMOVA based on COI + ND4 indicated that 36.1%, 11.73%, and 43.58% of the overall variation was among populations, and 63.9%, 88.27%, and 56.42% was within populations, of S. coreana, S. gottschei, and S. tegulata, respectively. For S. forticosta, 54.82% and 45.18% of the variation was among and within populations, respectively (Tables 2, 5, 8 and 11), in agreement with the minimum spanning network results. This indicates low genetic variation among geographic locations, except for S. forticosta populations, and suggests weak phylogeographic structures. In addition, the small inter-population variation values indicate restricted gene flow among regional populations. Inter-population genetic differentiation is affected by the blocking of gene flow by physical and physiological barriers, and by genetic drift and reduced species abundance (Jacquemyn et al., 2004). Wright (1965) reported that pairwise FST values of 0.05–0.15, 0.15–0.25, and > 0.25 indicate slight, considerable, and large genetic differentiation among populations, respectively. In this study, the average FST value was 0.139 for S. coreana, 0.106 for S. gottschei, 0.496 for S. forticosta, and 0.381 for S. tegulata, indicating high genetic variation and low genetic communication for S. forticosta and S. tegulata. The populations of semisulcospirids from different river systems showed significant genetic differentiation due to limited gene flow, which is in accordance with the findings of other studies (McGlashan & Hughes, 2000). However, only S. forticosta showed limited gene flow among populations and high pairwise FST values (0.314–0.624). The level of gene flow among local populations of the three other species was high, although distinct and rare haplotypes existed in different regions. The pairwise FST values of S. coreana ranged from 0.209 to 0.541 and 0.328 to 0.620 between the YD population and the other populations, and between the HY population and the other populations, respectively, suggesting a longer evolutionary history than the other regional populations. The pairwise FST values between the YP population and the other populations of S. gottschei were 0.168–0.253. In addition, the pairwise FST values between the CHU population and the other populations of S. tegulata, and between the YS population and the other populations of S. tegulata, were 0.143–0.634 and 0.303–0.913, respectively. These high genetic differentiation and low gene flow values are likely attributable to ecological factors, such as the sedentary lifestyle of the snail (Gu et al., 2015). However, the genetic distance and geographic distance of most populations of the two species were not correlated, indicating active gene flow. This was likely caused by transport by waterfowl, headwater capture, tectonic-driven changes in drainage patterns (Pfenninger et al., 2011) or marked larval dispersal (An et al., 2012). Similar results have been reported for other snail species, such as Bulinus truncatus (Zein-Eddine et al., 2017) and Juga (Strong & Whelan, 2019). Domestic drainage typically involves relatively short streams—with the obvious exceptions of the Geum river, Yeongsan/Seomjin river, Nakdong river, and Han river—and the populations in those streams showed various degrees of geographical genetic differentiation (Bae & Suk, 2015). The Korean inland endemic fish, Iksookimia pacifica, was collected in streams draining into the East Sea and clustered into a northern group and a southern group; however, gene flow was active in only one population in each of the northern and southern groups (Jang et al., 2017). This population was presented wahlund effect as the result of the change in biological distribution due to artificial disturbance by release the seed as a result of the wall effect (Jang et al., 2017). Melania snails must be released into the same water system from which their mothers were collected to prevent artificial disturbance of the ecosystem (Cho et al., 2013). However, S. coreana and S. gottschei collected nationwide had the lowest geographic genetic differentiation; this finding warrants further investigation.
Conclusion
We analyzed the genetic diversity and population structure of four Semisulcospira species using mtDNA markers. In summary, a high level of haplotype diversity and low level of nucleotide diversity were detected among the 18 populations of Semisulcospira. Therefore, the drastic decline in drainage in the ROK due to over-harvesting has not resulted in low genetic diversity. Although the populations are expanding, this species requires management and conservation because of its restricted distribution, endemism, and consumption by humans. Through continuous genetic diversity analysis, it is necessary to explore the direction of resource management for populations where regional genetic differences appear. This study indicates that the domestic species has gone genetic diversion in each water system, especially in S. forticosta. Therefore, release of population with genetic difference for resource restoration should be done with special care to conserve the unique genetic population in each water system.