






Introduction
The order Octopoda has two main distinguished suborders, Cirrata and Incirrata. Cirrata possess rows of cirri, while Incirrata are devoid of cirri. The Incirrata suborder consists of two superfamilies: Argonautoidea and Octopodoidea. Tremoctopus is a genus of Tremoctopodidae, which belongs to the Argonautoidea superfamily. Tremoctopus is commonly known as the blanket octopus and contains four species (Finn, 2016): Tremoctopus violaceus, Tremoctopus gracilis, Tremoctopus robsoni, and Tremoctopus gelatus. T. violaceus was previously classified into two subspecies: Tremoctopus violaceusviolaceus and Tremoctopus violaceus gracilis (Thomas, 1977). However, the two subspecies are now treated as distinct species, T. violaceus and T. gracilis (Finn, 2016).
In East Asia, the presence of T. violaceus has been recorded in the coastal waters of Japan and the nearby South China Sea (Georeferenced records in GBIF, https://www.gbif.org), as well as on the shore of Samcheok-si, Kangwon-do, South Korea in 2019. Specimens of T. violaceus are very rarely collected in East Asia; therefore, its distribution status, dispersing history, and phylogenetic relationships with populations found in other regions are unclear.
Ever since the mitochondrial (mt) genome of Octopus vulgaris was first reported (Yokobori et al., 2004), complete mt genomes of over 20 Octopoda species have been reported. Most animal mt genomes generally have a compact size (14–19 kb), circular form, and comprise 13 protein-coding genes (PCGs), 22 tRNAs, two rRNAs, and a noncoding control region (Boore, 1999). The mt genome has been used for identifying species, tracking evolutionary history, and making inferences related to comparative genomics from nucleotide composition, structural features, and gene rearrangement, etc. (Boore, 1999; Kumazawa et al., 1996; Li et al., 2016; Yu et al., 2019). Molecular phylogeny performed using mt genomes is useful for resolving controversial evolutionary relationships among various animal groups. Uribe & Zardoya (2017) revisited the phylogeny of Cephalopoda using complete mt genomes, and suggested that Octopus and Octopodidae are polyphyletic, as reported in previous studies (Cheng et al., 2013; Magallón-Gayón et al., 2020).
Recently, the importance of biodiversity has led to worldwide attention because biodiversity affects ecosystem functions (Baert et al., 2018; Isbell et al., 2018; O’Connor et al. 2017). Biodiversity is highly threatened by climate change, therefore, many advanced countries are making a lot of efforts to preserve global biodiversity through the Nagoya protocol, etc. Research on biodiversity begins with identifying species, and genetic analysis methods are widely used as a tool to identify species and analyze phylogenetic relationships (Chen & Wang, 2021; Yi et al., 2021).
This study presents the sequencing of the complete mt genome of T. violaceus and the description of the T. violaceus mt genome organization and phylogenetic analysis using the mt 13 PCG sequences that are generally found in species of the Octopoda.
Materials and Methods
Our study was performed using a T. violaceus specimen caught by a fisherman in the sea near the Jeju east pier, and donated for research. Genomic DNA was extracted from a small piece of muscle using a Nucleospin Tissue Kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s protocol. The complete mt genome was amplified and sequenced using newly designed primers, including nested primers for primer walking sequencing. All sequencing reactions were conducted at Bionics (Seoul, Korea).
The complete mt genome was annotated using MITOS (Bernt et al., 2013) and ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder) using the invertebrate mitochondrial code. The complete mt genome map was drawn using SnapGene 5 software. The codon usage of 13 PCGs was analyzed using MEGA X (Kumar et al., 2018). The tRNA genes were identified using web-based tRNAscan-SE software (Lowe & Eddy, 1997) and ARWEN (Laslett & Canbäck, 2008).
To reconstruct the phylogenetic tree within the Octopoda, previously reported mt genomes of species in Octopoda were collected from the GenBank database and 13 PCG sequences were aligned using Clustal X (Thompson et al., 1997) with default settings. Bayesian inference (BI) performed using MrBayes 3.2 software (Ronquist et al., 2012) was used for phylogenetic tree reconstruction. The GTR + G + I model was selected as the best evolutionary model using jmodeltest2 (Darriba et al., 2012). Four Markov chains were run for 100,000 generations and sampled every 100 generations to obtain a posterior probability (PP) distribution of 1,000 trees. Vampyroteuthis infernalis (AB266515) was used as the outgroup (Uribe & Zardoya, 2017).
Results
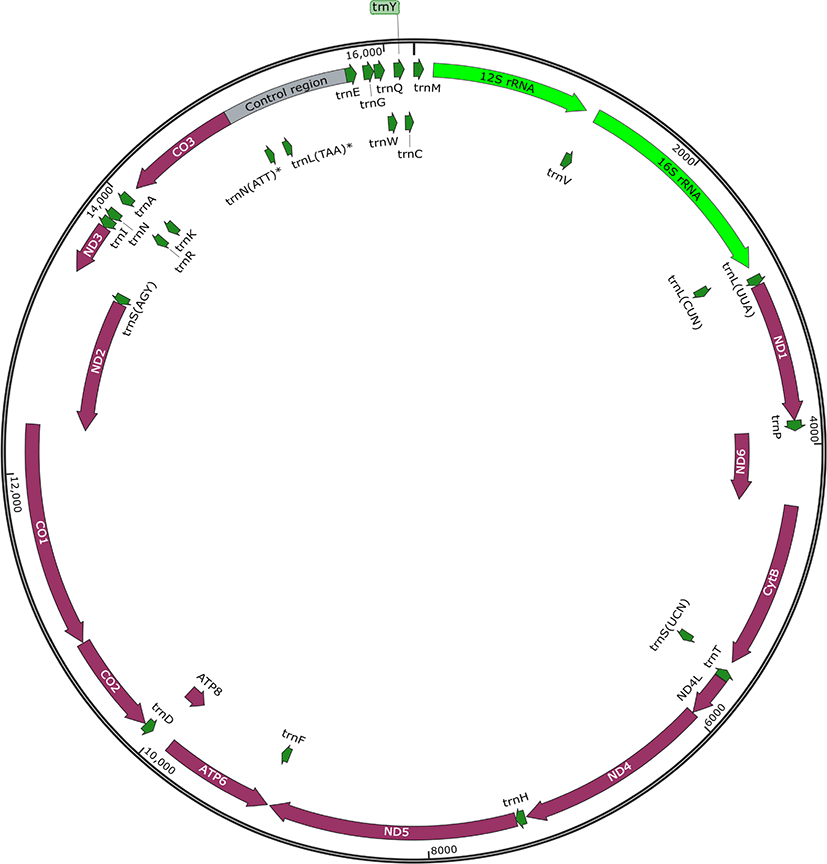
The complete mt genome sequence of T. violaceus was 16,191 bp long (GenBank accession No. MZ043857). The content of A, C, G, and T was 31.68 % (5,130/16,191), 7.71 % (1,248/16,191), 20.02 % (3,242/16,191), and 40.58 % (6,571/16,191), respectively. The mt gene order was identical to that of other Octopoda species.
As observed in other animals, the mt genome of T. violaceus has 13 PCGs encoded on the heavy and the light strand. NADH dehydrogenase subunit 1 (ND1), ND6, cytochrome b, ND4L, ND4, and ND5 were encoded on the heavy strand and the other genes were encoded on the light strand (Fig. 1). On the heavy strand, ND6 and CytB genes overlapped by 8 nucleotides, while ATPase subunit 8 and 6 genes overlapped by eight nucleotides. On the light strand, cytochrome c oxidase subunit (CO) 1 and ND2 genes overlapped by 23 nucleotides. All PCGs were initiated with the ATG codon, excluding ND4 and ATP6, that were initiated with ATC and ATT, respectively, and terminated with TAG, TAA, TA, or T (Table 1). Among the 13 PCGs, only two PCGs, ND1, and ND5, terminated with the incomplete stop codons, T and TA, respectively.
The total number of codons used in the 13 PCGs was 3,750, wherein codons corresponding to trnI were the most used (257 times), while codons for trnR were used the least (three times) (Table 2). Among the 3,750 codons, the proportion of codons for trnL was 15.2 % (571/3,750, the most used codon), whereas that for trnR was 1.3 % (50/3,750, the least used codon). trnL and trnS were coded by six and eight different codons, respectively. trnA, trnR, trnG, trnP, trnT, and trnV were coded by four different codons, while the other sequences were coded by two different codons.
The 12S and 16S rRNA genes of the T. violaceus mt genome were 1,077 bp and 1,472 bp in length, respectively (Table 1). As observed in other Octopus genomes, these two rRNA genes were located between trnM and trnL, and were separated by trnV.
The mt genome of T. violaceus was seen to have 24 tRNAs, whereas the mt genomes of most animals have 22 tRNAs. The two extra tRNAs, found located in the control region, were trnN and trnL (Fig. 1). The secondary structure of both additional tRNAs showed an abnormal cloverleaf structure without the D-arm (Fig. 2).

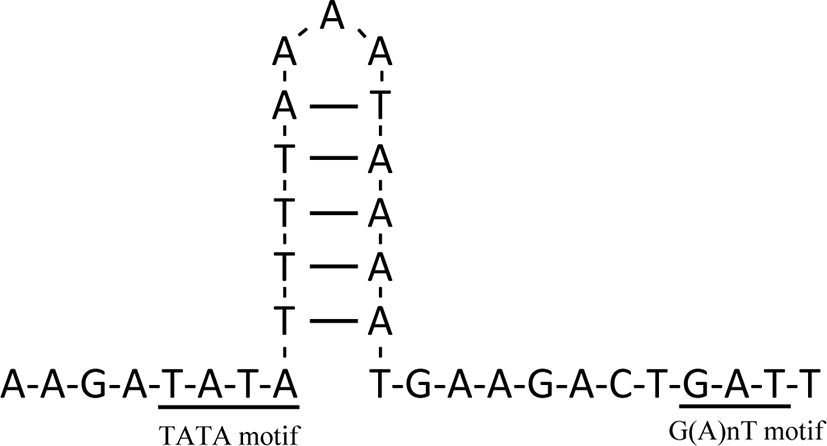
The mt genome of T. violaceus had four noncoding regions, including the control region known as the D-loop. The control region, a major noncoding region, was 855 bp long and located between CO3 and trnE, while other noncoding regions — 1, 2, and 3 — were 66 bp, 39 bp, and 48 bp, respectively (Table 1). Similar to the mt genomes in other animals, the control region of the T. violaceus mt genome showed high A + T content (83.16 %), and had a stem-loop structure flanked by two elements (TATA motif and G(A)nT motif) (Fig. 3).
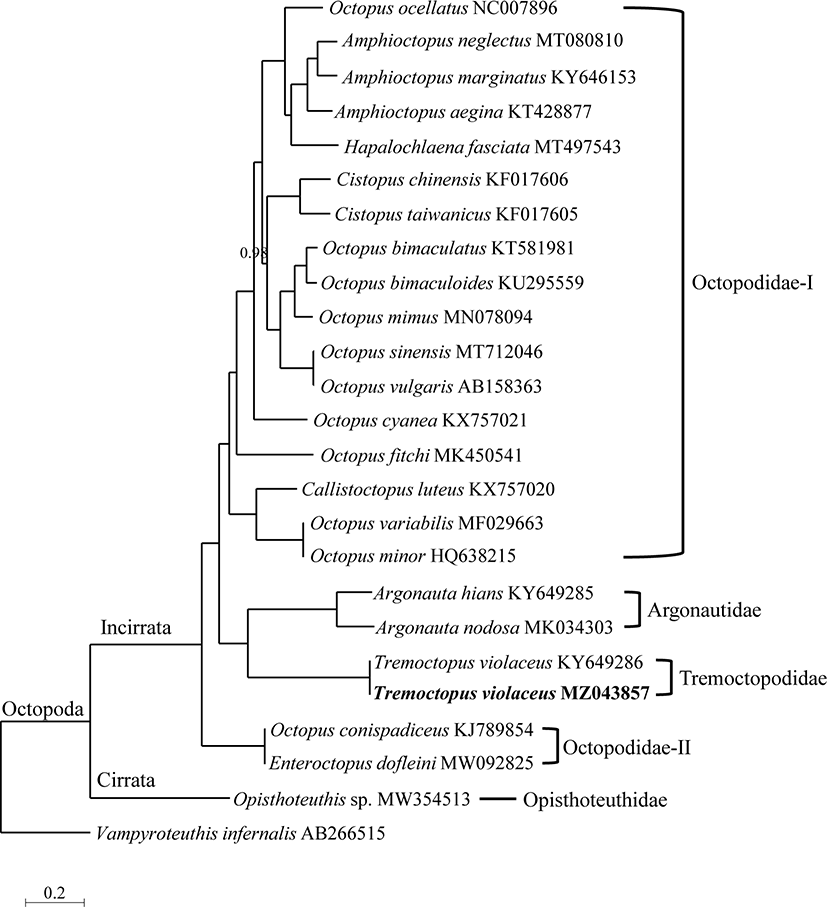
In this study, nine genera from four families registered in the GenBank database were analyzed for obtaining Octopoda phylogeny. BI using 13 PCGs showed that the Tremoctopodidae family was supported as a sister group to Argonautidae (Fig. 4). The branches of Tremoctopodidae and Argonautidae were located in the basal position of the Incirrata clade. The phylogenetic tree showed Octopus and Octopodidae to be polyphyletic. Octopus species were seen to be dispersed around the phylogenetic tree and the Octopodidae family was divided into two clades, Octopodidae-I and Octopodidae-II. Octopodidae-II was the first to branch from the Incirrata clade, and Tremoctopodidae and Argonautidae branched later.
Discussion
The general features of the mt genome were identical to those of other Octopus mt genomes. ND6 and CytB genes, as well as the ATPase subunit 8 and 6 genes had an overlap of eight nucleotides, while CO1 and ND2 genes had an overlap of 23 nucleotides. These overlapping features are common and have been found in the mt genomes of other octopuses (Cheng et al., 2013; Chiu et al., 2018; Tang et al., 2018). The alternative initiation codons, AUA, AUU, ATC, GUG, and UUG have been reported in invertebrate mt genomes (Donath et al., 2019). Among these, ATC and ATT were used in ND4 and ATP6, respectively, in the T. violaceus mt genome. Meanwhile, incomplete termination codons, T and TA, were used in ND1 and ND5, respectively. Incomplete termination codons are very common in various animal mt genes, including those of mammals, fishes, reptiles, amphibians, and invertebrates (Cui et al., 2007; Kumazawa et al., 1996; Oh & Jung, 2019; Oh et al., 2019; Sumida et al., 2001). Incomplete termination codons are said to be completed via posttranscriptional polyadenylation (Ojala et al., 1981).
Although an animal mt genome normally has 22 tRNAs, the T. violaceus mt genome was seen to have 24 tRNAs. The additional tRNAs, trnN and trnL, were located in the noncoding control region (Fig. 1). Additional tRNAs in the Octopoda mt genome have been reported in Cistopus chinensis (Cheng et al., 2013). Both extra tRNAs have D-armless structures; abnormal secondary structures of some tRNAs in the mt genome have often been identified in various animals (Danic-Tchaleu et al., 2011; Jarošová et al., 2016; Morrison, 2010; Pons et al., 2019; Yokobori et al., 2005). Although the occurrence of extra tRNAs, and their functional mechanisms, in the animal mt genome remain unclear, they might share the same amino acid with conventional tRNAs, as observed in the ascidian mt genome (Kondow et al., 1999).
Four noncoding regions, including the control region, were identified in the mt genome of T. violaceus. Meaningful structural features were absent in the three noncoding regions other than the control region. Generally, the control region is believed to be involved in DNA replication and transcriptional regulation (Clayton, 1982). The control region of the T. violaceus mt genome has a stem-loop structure and two elements that are presumed to be associated with the processes of replication and transcriptional regulation (Wei et al., 2010). Although the TATA motif and G(A)nT motif have been reported in other invertebrates (Kuhn et al., 2008; Schultheis et al., 2002), this study is the first to report these elements in Octopoda.
The Tremoctopodidae, which was well supported as a sister group to the Argonautidae, was seen to consist only one genus, Tremoctopus, and the branches of both families were located at the basal position in Incirrata (Fig. 4). Consequently, Tremoctopus was considered to have diverged relatively earlier within the Octopoda.
Our BI tree showed the polyphyly of Octopus and Octopodidae, which has been supported by molecular data from previous studies (Carlini et al., 2001; Cheng et al., 2013; Chiu et al., 2018; Magallón-Gayón et al., 2020; Uribe & Zardoya, 2017). Our BI tree showed Octopodidae to be divided into two clades (Fig. 4). Octopodidae-I consisted of all species in Octopodidae, excluding Octopus conispadiceus and Enteroctopus dofleini, which were found in Octopodidae-II. Octopus ocellatus, Octopus variablilis, Octopus minor, and Octopus conispadiceus require taxonomic reconsideration (Cheng et al., 2013; Uribe & Zardoya, 2017).
In this study, we described the complete mt genome of T. violaceus and showed its phylogenetic status. In this study, the two putative extra tRNAs that were found represented a rare feature observed in Octopoda, and, for the first time, a stem-loop structure with two elements was reported in Octopoda. Phylogenetic analysis showed the polyphyly of Octopus and Octopodidae, and the relatively early divergence of the Tremoctopus genus. Our data provides useful information for studying comparative mt genomics and phylogenomics of Tremoctopus, and further facilitates evolutionary studies of Octopoda.
Genetic analysis is very useful as a tool to measure biodiversity levels in each ecosystem, and sufficient genetic data can help determine the priorities of biodiversity conservation organisms. The mt genome sequence is also used in many studies for speciation, gene flow, population dynamics, etc. Therefore, the mt genome will provide useful information for establishing biodiversity conservation strategies.