






Introduction
Hepatocellular carcinoma (HCC) is a malignant tumor that originates in the liver and exhibits the third- highest mortality rate worldwide. Notably, the development of HCC is promoted by risk factors such as chronic hepatitis B and C, alcoholic and chronic liver disease, etc. (McGlynn et al., 2021; Rinaldi et al., 2021). Despite specific causes and advances in modern medical technology, the incidence and mortality of HCC remain unresolved. In particular, chemotherapy is limited by cost, drug resistance, and side effects; thus, an efficient treatment regimen needs to be established (Chakraborty & Sarkar, 2022; Wakabayashi et al., 2019).
Auranofin is a gold compound known as US Food and Drug Administration (FDA)-approved treatment for rheumatoid arthritis (Roder & Thomson, 2015). Auranofin is a thioredoxin reductase (TrxR) inhibitor, which acts as a regulator of cellular redox, cell proliferation, and a component of antioxidant systems (Abdalbari & Telleria, 2021). Cui et al. (2022) have demonstrated that auranofin can induce apoptosis in human lung cancer cells by increasing the accumulation of reactive oxygen species (ROS) via inhibition of TrxR activity and glutathione depletion. Furthermore, auranofin exerts anticancer activity by inactivating extracellular signal-regulating kinase and protein kinase B (Akt), accompanied by mitochondrial dysfunction through the modulation of B-cell lymphoma 2 (Bcl-2) family proteins (Wen et al., 2019). Accordingly, auranofin could be an effective component of various anticancer treatment strategies by generating ROS and mitochondrial dysfunction in diverse cancer cells (Abdalbari & Telleria, 2021; Massai et al., 2022). Moreover, recent studies have reported the synergistic effect of auranofin for maximizing anticancer activity, suggesting its potential as an effective anticancer agent (Han et al., 2019; Ye et al., 2019; Zhu et al., 2020).
Marine algae contain a high polyamine content and can exhibit various physiological activities (Igarashi & Kashiwagi, 2019; Puleston et al., 2019). Spermidine, a type of polyamines, is widely distributed in living organisms and is also found in some red and brown algae (García-Jiménez et al., 2007; Kumar et al., 2015). Spermidine, synthesized by putrescine and S-adenosylmethionine, exhibits multiple biological functions, including cell differentiation, proliferation, angiogenesis, gene expression, anti-aging, anti-inflammatory, and anticancer activities (Madeo et al., 2018; Pegg, 2014). Specifically, spermidine can prevent sepsis-induced acute kidney injury by downregulating the Nod-like receptor family pyrin domain containing 3 inflammasomes and interleukin-1β in macrophages (Li et al., 2022). Furthermore, spermidine can improve the function of human cells and delay aging (Madeo et al., 2018; Wang et al., 2020; Xu et al., 2020). In addition, oral spermidine can suppress HCC and liver fibrosis in rats by autophagy (Yue et al., 2017), suggesting its potential as a promising candidate for cancer treatment.
To overcome the limitations of HCC therapy, the present study aimed to evaluate the synergistic effect of auranofin and spermidine as an effective combination therapy for HCC treatment. In addition, we investigated the mechanism underlying the synergistic anticancer activity mediated by auranofin and spermidine, providing evidence to support their potential as a therapeutic strategy with few side effects.
Materials and Methods
Auranofin, spermidine, dimethyl sulfoxide (DMSO), crystal violet, 2’,7’-dichlorodihydrofluorescein diacetate (H2DCF-DA), N-acetylcysteine (NAC), and 1,1’,3,3’-Tetraethyl-5,5’,6,6’-tetrachloroimidacarbocyanine iodide (JC-1) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), penicillin-streptomycin, fetal bovine serum (FBS), and trypsin-ethylenediaminetetraacetic acid (EDTA) were obtained from Welgene (Daegu, Korea). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), enhanced chemiluminescence (ECL) solution, and MitoTrackerTM Red were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Formaldehyde was purchased from Junsei (Tokyo, Japan). The fluorescein isothiocyanate (FITC) annexin V apoptosis detection kit, CycletestTM plus DNA reagent kit, and skim milk were obtained from BD Biosciences (Franklin Lakes, NJ, USA). Nitrocellulose membranes were purchased from GE Healthcare (Chicago, IL, USA). Anti-poly (ADP-ribose) polymerase (PARP), Bcl-2, Bax, Akt, and secondary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). β-actin was purchased from Bioworld Technology (Nanjing, China). Phosphatidylinositol-3 kinase (PI3K), phosphorylated PI3K, and phosphorylated Akt were obtained from Cell Signaling Technology (Danvers, MA, USA).
Hep3B cells, a human HCC cell line (American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM containing 1% penicillin-streptomycin and 10% FBS in an incubator with 5% CO2 at 37°C. Auranofin was dissolved in DMSO at 1 mM, and spermidine was dissolved in water at 1 M, followed by storage at –20°C until use. Cells were treated with 1 μM auranofin and different spermidine concentrations for 24 h. After treatment, cells were used for further experiments.
Following treatment with auranofin and spermidine, changes in cell viability were measured using the MTT assay. In brief, cells were treated with 1 μM auranofin and various spermidine concentration (20, 40, and 60 μM) for 24 h. The incubated cells were treated with 0.5 mg/mL MTT solution and reacted for 2 h at 37°C. After the reaction, the generated formazan crystals were dissolved in DMSO and the absorbance was measured at 540 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cells were seeded in 6-well plates (1,000 cells/well), followed by treatment with auranofin and spermidine for another 24 h. Next, cells were washed with phosphate-buffered saline and cultured for 10 days in fresh medium. After incubation, cells were fixed with 4% formaldehyde and then stained with 0.05% crystal violet. The stained colonies were observed under a microscope (Carl Zeiss, Oberkochen, Germany) at Core-Facility Center for Tissue Regeneration, Dong-Eui University.
Apoptosis was quantitatively assessed using annexin V-positive cells and cell cycle analyses. Accordingly, the cells with auranofin and spermidine were harvested using trypsin-EDTA. The annexin V/PI assay was performed using the FITC annexin V apoptosis detection kit, and cell cycle analysis was performed using the CycletestTM plus DNA reagent kit. Stained cells were analyzed using an Accuri C6 flow cytometer (BD Biosciences).
JC-1 dye was used to examine the loss of MMP (Δψm). In brief, cells treated with auranofin and spermidine were harvested and stained with JC-1 dye (10 μM) for 20 min. After incubation, fluorescence intensity was detected using an Accuri C6 flow cytometer (BD Biosciences).
Cells were lysed with lysis buffer (250 mM NaCl, 25 mM Tris-Cl [pH 7.5], 5 mM EDTA [pH 8.0], 1% NP-40, 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 5 mM dithiothreitol, and protease inhibitor cocktail) to extract total proteins. The amount of protein in all samples was quantified at 3 μg/μL, separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis, and then transferred to nitrocellulose membranes. Membranes were blocked with 5% skim milk and reacted with primary antibodies overnight at 4°C. Subsequently, the proteins on the membrane were treated with the appropriate secondary antibody and an ECL solution, and the bands were detected using a chemiluminescence system (Fusion Solo S system, Vilber Loumat, Collégien, France).
After treating with auranofin (1 μM) and spermidine (60 μM) for 1, 3, 6, and 24 h, the cells were immediately stained with H2DCF-DA dye to determine intracellular ROS for 15–20 min. The samples were then analyzed using a flow cytometer and examined by fluorescence microscopy (Thermo Fisher Scientific). The mitochondria were labeled using MitoTracker Red to determine the location of ROS generation. NAC was used as a positive control to measure ROS levels.
All data are expressed as mean ± SD and statistical analyses were performed using Graphpad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Analysis of variance was used to compare the experimental groups, and Tukey’s post hoc test was performed to determine statistical significance (p < 0.05, p < 0.01, and p < 0.001).
Results
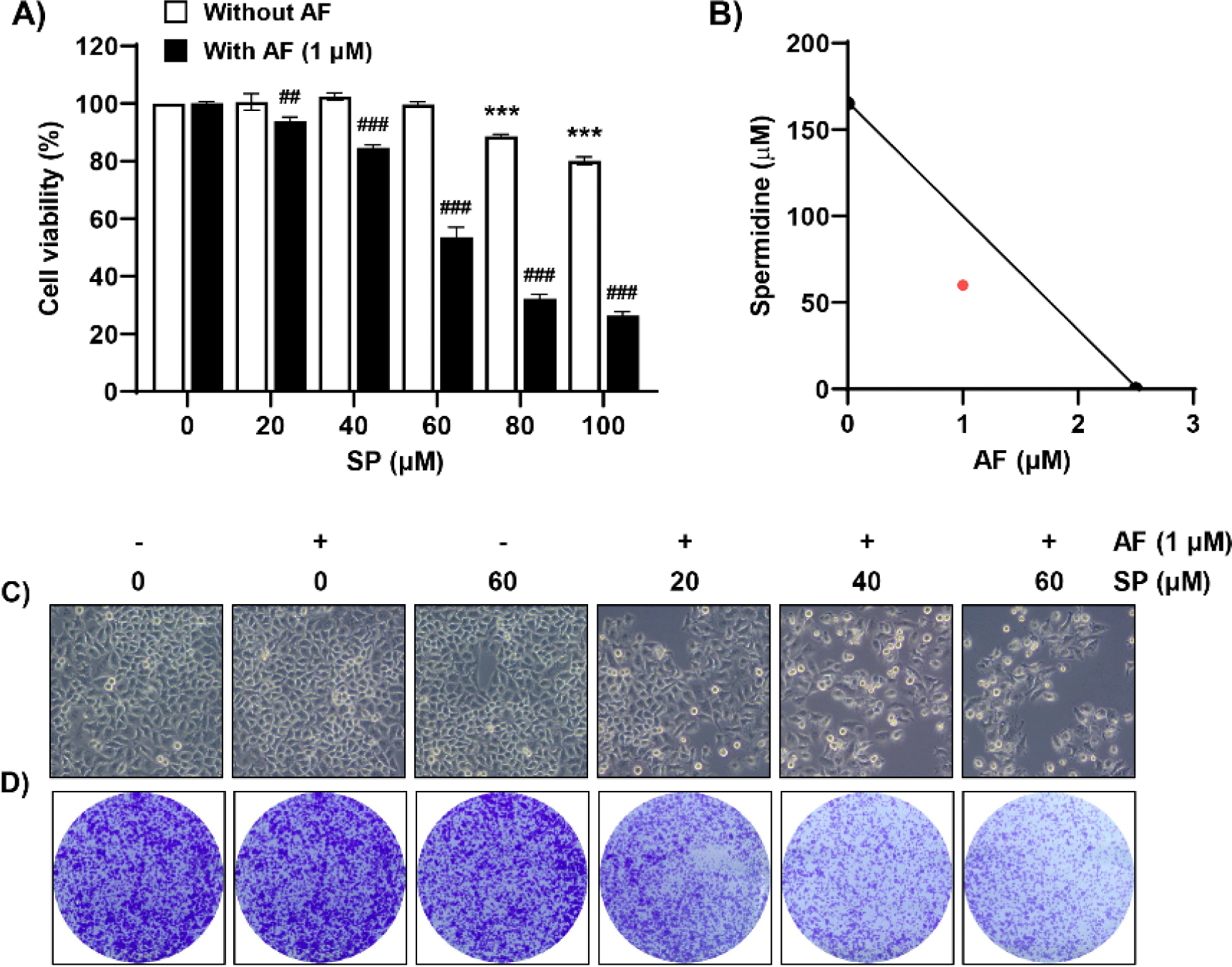
Herein, we investigated the effect of auranofin on the spermidine-induced decrease in Hep3B cell proliferation. As shown in Fig. 1A, auranofin significantly accelerated the spermidine-induced decrease in cell viability. The synergistic effect of two drugs on the suppression of cell proliferation was confirmed using isobologram analysis (Fig. 1B). Furthermore, we examined cell morphology and colony formation to examine cell density and colony formation ability, which were reduced in the presence of auranofin in a spermidine concentration-dependent manner (Fig. 1C and 1D). Therefore, these results indicated that co-treatment with auranofin and spermidine affords a synergistic effect on the viability of Hep3B cells.
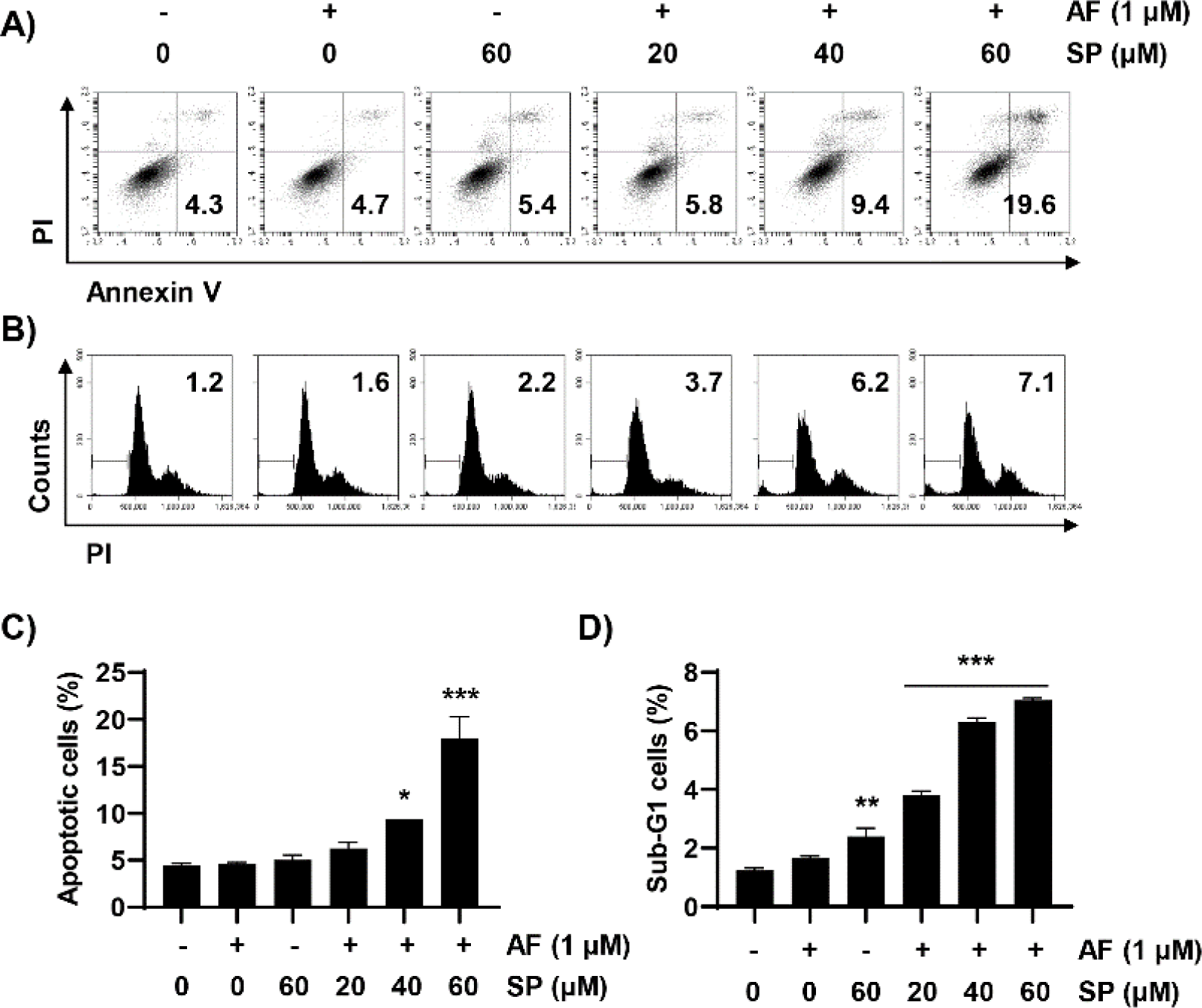
To examine whether reduced cell viability induced by auranofin and spermidine could be attributed to apoptosis, we measured the frequencies of annexin V-positive cells and the sub-G1 phase using flow cytometric analysis. We observed that combined treatment with auranofin and spermidine increased the frequency of annexin V-positive cells, that is, apoptosis-induced cells (Fig. 2A and 2C). Additionally, cell cycle analysis was performed to measure the fractional DNA content. Auranofin and spermidine treatment increased the percentage of cells remaining in the sub-G1 phase (Fig. 2B and 2D).
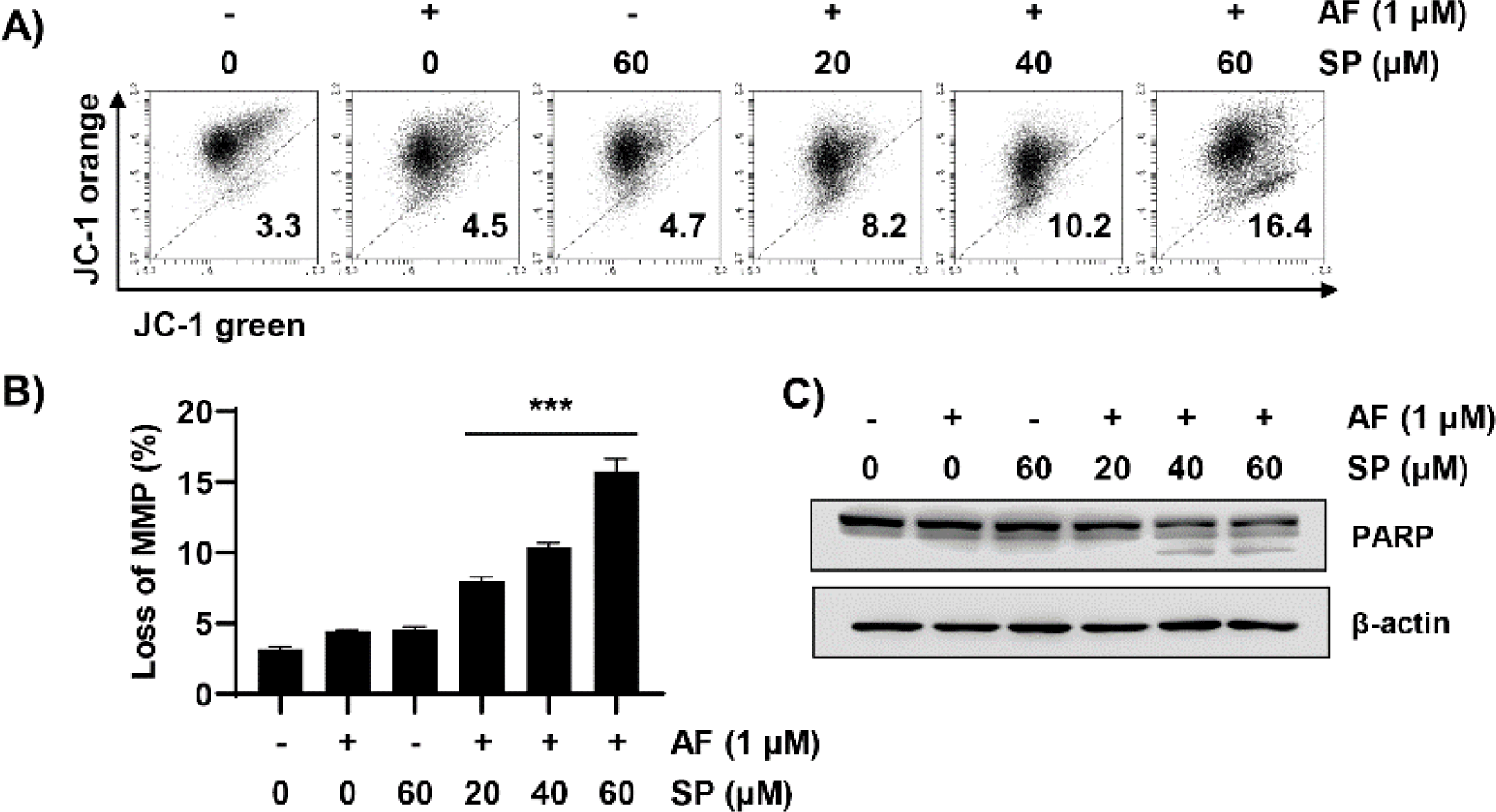
Next, we measured MMP (Δψm) loss using JC-1 dye to quantitatively evaluate mitochondrial changes during auranofin and spermidine-induced apoptosis. These results indicated that combined treatment with auranofin and spermidine significantly increased MMP (Δψm) loss (Fig. 3A and 3B). Moreover, combination treatment with auranofin and spermidine increased the expression of the cleaved form of PARP, an apoptosis marker (Fig. 3C). Overall, these results indicated that auranofin and spermidine-induced apoptosis involves mitochondrial involvement.
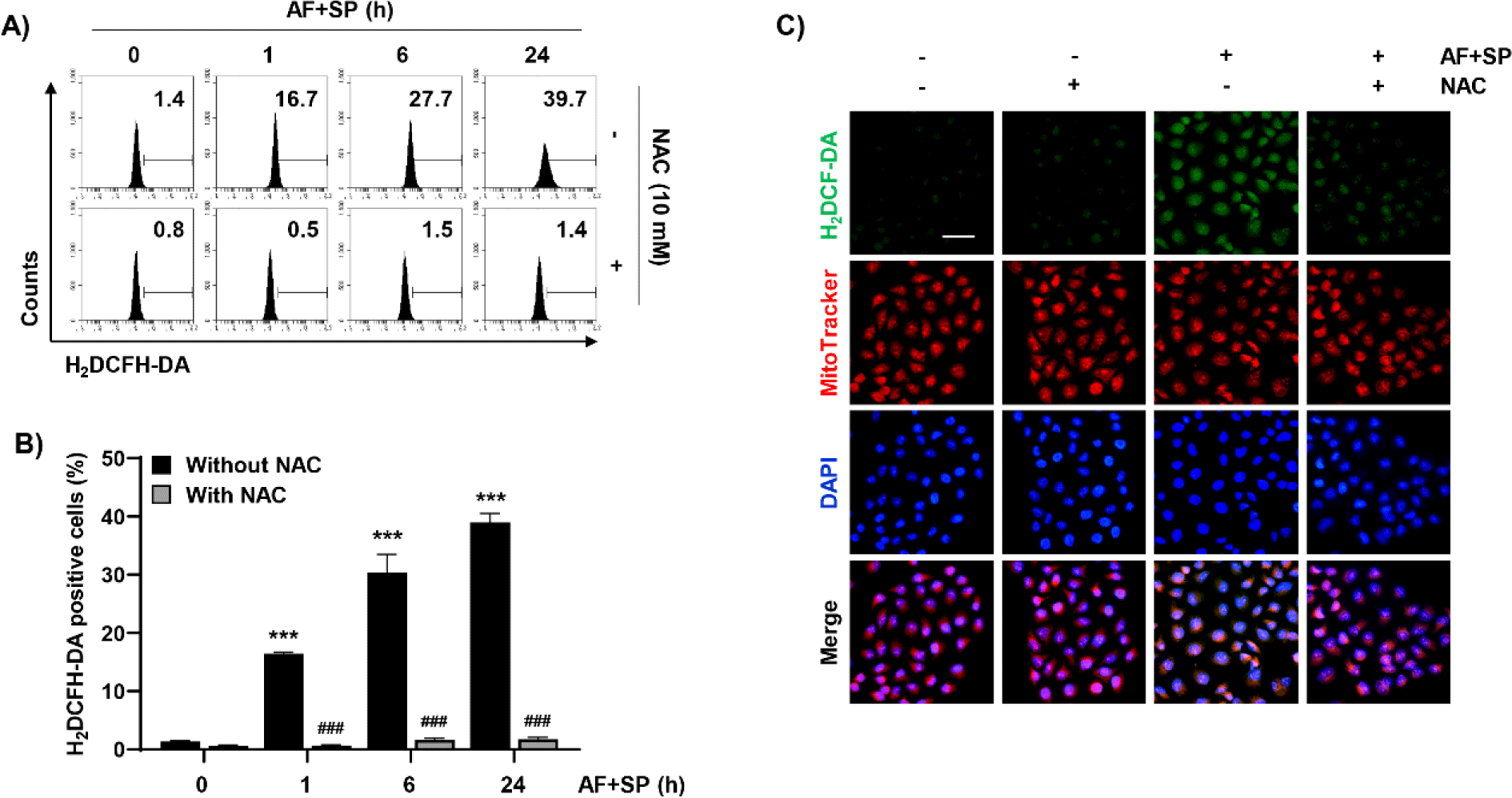
Given that intracellular ROS plays a key role in apoptosis induction, ROS levels generated following treatment with auranofin and spermidine were measured using the H2DCF-DA dye. To measure intracellular ROS generation, the effects of auranofin and spermidine treatment were evaluated at different time points. ROS generation increased with increasing exposure time to auranofin and spermidine. However, pretreatment with NAC, a ROS scavenger, dramatically decreased the increased ROS levels induced by treatment with auranofin and spermidine (Fig. 4A and 4B). Furthermore, fluorescence microscopy results revealed that H2DCF-DA (green fluorescence) was increased by auranofin and spermidine, which was co-localized with MitoTracker Red, a dye used to stain mitochondria in proportion to their membrane potential (Fig. 4C).
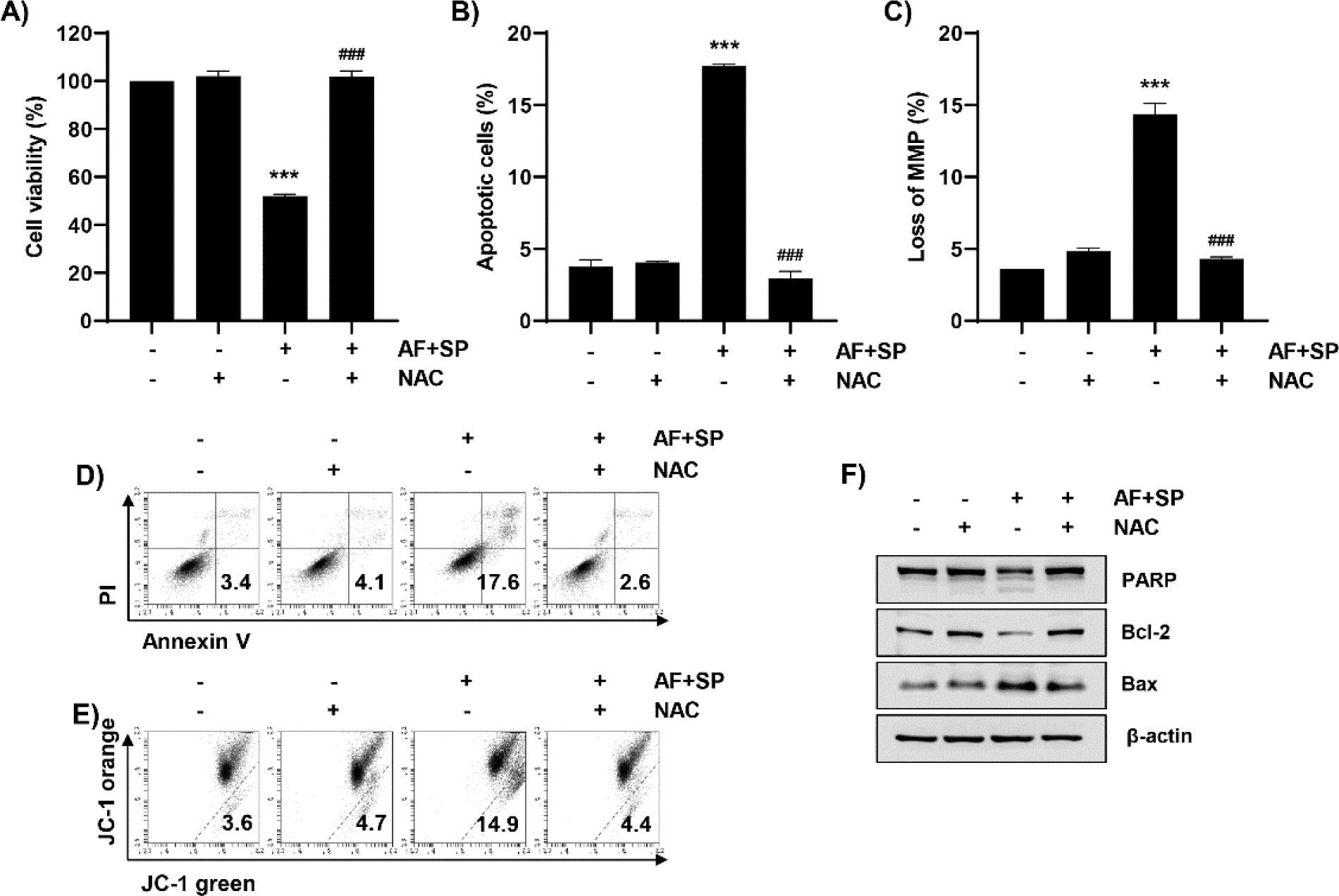
We next examined changes in apoptosis induced by auranofin and spermidine under suppressed ROS generation. As indicated in Fig. 5A, 5B, and 5D, auranofin and spermidine-induced growth suppression and apoptosis were completely blocked by NAC. Moreover, mitochondrial dysfunction increased by auranofin and spermidine was recovered to control levels by NAC (Fig. 5C and 5E). Therefore, we further examined the expressions of Bcl-2 and Bax, which play key role in mitochondria-mediated apoptosis, using Western blot analysis. As indicated in Fig. 5F, NAC could restore the auranofin and spermidine-induced increase in Bax expression and decrease in Bcl-2 expression (Fig. 5F). Moreover, NAC reduced cleaved PARP levels induced by auranofin and spermidine.
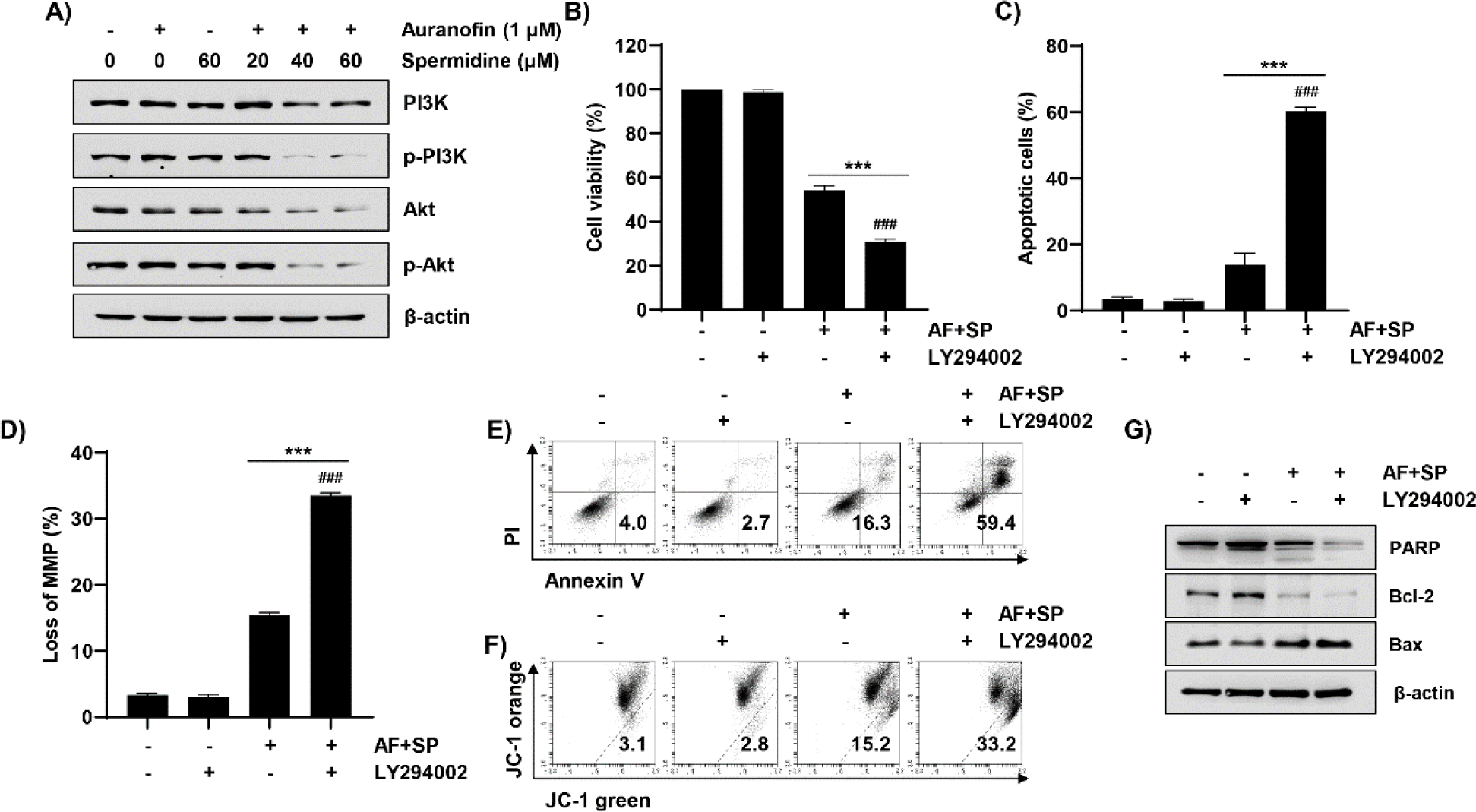
As shown in Fig. 6A, treatment with auranofin and spermidine decreased expressions levels of phosphorylated forms of PI3K and Akt. Therefore, we used the PI3K inhibitor to establish the role of the PI3K/Akt signaling pathway in auranofin- and spermidine-induced apoptosis. Based on the cell viability and flow cytometry results, we observed that LY294002, a PI3K inhibitor, aggravated cell growth inhibition, apoptosis, and MMP (Δψm) loss induced by auranofin and spermidine (Fig. 6B–6F). Additionally, LY294002 aggravated the expressions of apoptosis-related proteins, which were altered by auranofin and spermidine. As shown in Fig. 6G, the total form of PARP and the expression of Bcl-2 were further decreased, and the expression of Bax was markedly increased following LY294002 treatment when compared with auranofin and spermidine treatment.
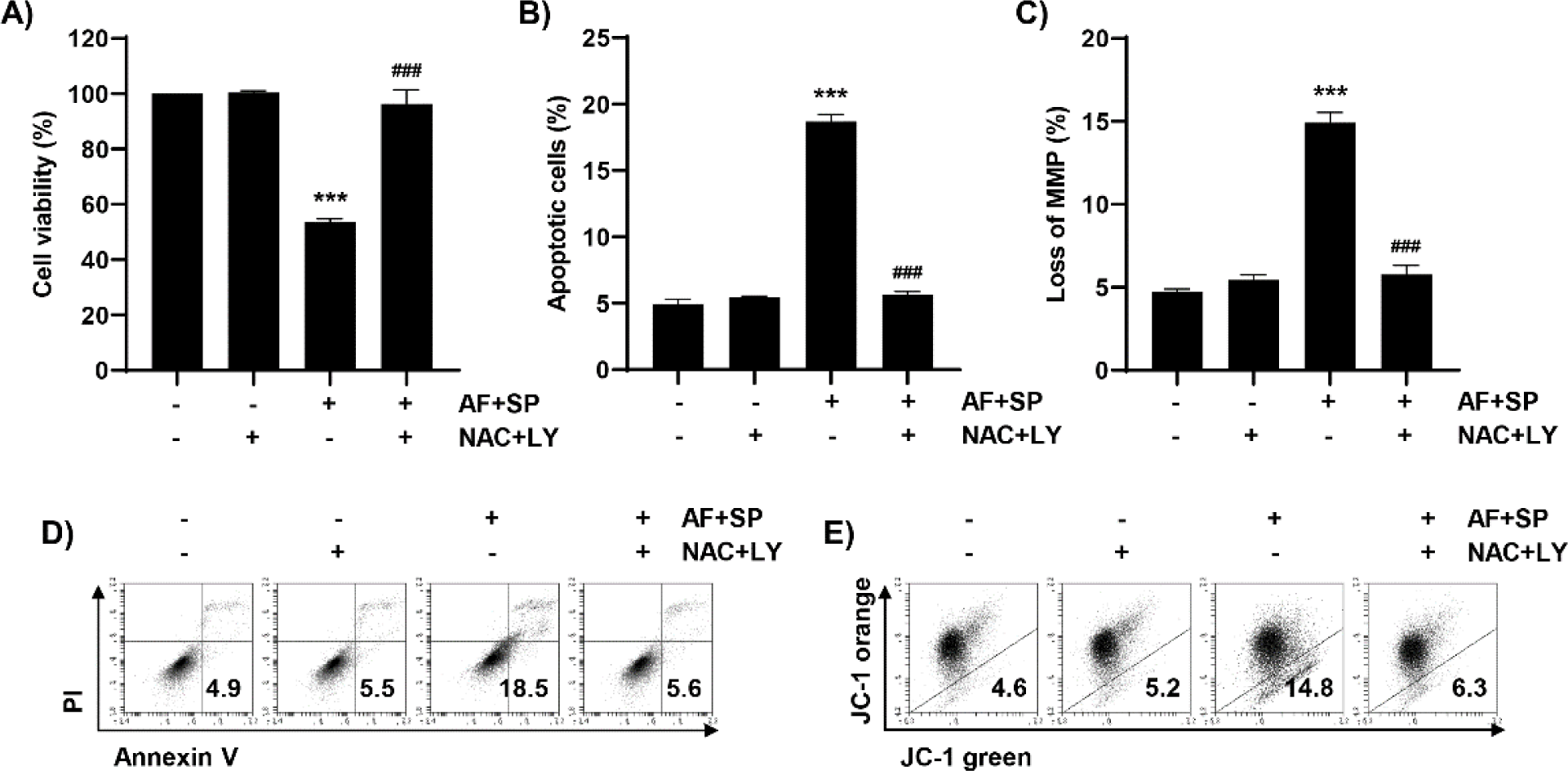
Considering auranofin- and spermidine-induced apoptosis, we aimed to determine the role of the ROS and PI3K/Akt signaling pathways. Accordingly, changes in apoptosis induced by auranofin and spermidine were investigated by using ROS scavenger and PI3K inhibitor. As shown in Fig. 7A, NAC plus LY294002 could restore the reduced cell viability induced by auranofin and spermidine. Based on flow cytometry results, treatment with NAC plus LY294002 could reduce apoptosis and MMP loss induced by auranofin and spermidine (Fig. 7B–7E). Consequently, auranofin and spermidine induced apoptosis in Hep3B cells through the PI3K/Akt signaling pathway under ROS regulation.
Discussion
Treatment options for HCC include liver transplantation, resection, radiation therapy, and chemotherapy (Chakraborty & Sarkar, 2022; Foerster et al., 2022). However, effective strategies for treating HCC are yet to be established, given the poor prognosis owing to recurrence and rapid disease progression (Cucarull et al., 2022; El Dika et al., 2021). Considering chemotherapy, developing new drugs and identifying effective and safe therapeutic agents remain crucial to overcome disadvantages, such as low response rate, toxicity, and drug resistance (Chakraborty & Sarkar, 2022; Chen et al., 2019). Combination therapy is an efficient strategy to improve the treatment response and minimize drug resistance and side effects in oncology (Fisusi & Akala, 2019; Plana et al., 2022). Therefore, in the present study, combination therapy was suggested as a promising HCC treatment strategy, and auranofin and spermidine were selected as drugs for combination therapy.
Cancer treatment targeting the developed antioxidant system in cancer cells is a promising and efficient approach (Moloney & Cotter, 2018; Prasad et al., 2017). Auranofin exerts anticancer activity by inducing oxidative stress, which increases ROS generation through TrxR inhibition (Cui et al., 2022; Hwang-Bo et al., 2017). Spermidine, selected as the second drug in the combination therapy, is a polyamine abundant in seaweed and exerts various biological functions (Kumar et al., 2015). Therefore, spermidine is considered an appropriate drug for treating cancer cells without inducing side effects on normal cells while maximizing the anticancer activity of auranofin.
Our results demonstrated that the cell viability of Hep3B cells was suppressed by high-concentration of spermidine. Interestingly, combined treatment with auranofin and spermidine significantly decreased cell viability at low concentration of spermidine. Considering the isobologram plotted based on cell viability results, auranofin and spermidine acted synergistically. Additionally, treatment with auranofin and spermidine alone did not alter cell density and colony formation, but combined treatment could markedly reduce these parameters. These results support the synergistic effect of auranofin and spermidine on the anticancer activity in HCC. Apoptosis is a programmed cell death that has been actively examined to establish anticancer activity, and is characterized by DNA cleavage at regular intervals, nuclear condensation, caspase activation, and externalized phosphatidylserine that binds to annexin V (Elmore, 2007). Auranofin and spermidine can reportedly induce cancer cell apoptosis (Abdalbari & Telleria, 2021; Yue et al., 2017). Herein, we found that combined treatment with auranofin and spermidine could increase the frequency of apoptosis-positive cells and cells in the sub-G1 phase, both hallmarks of apoptosis.
Mitochondrial changes can profoundly impact the apoptosis process; in particular, disruption of MMP (Δψm) is closely related to the intrinsic apoptosis pathway. In addition, Bcl-2 family proteins control the permeability of the mitochondrial membrane, thereby playing an important role in intrinsic apoptosis (Dadsena et al., 2021; Warren et al., 2019). Our findings revealed that auranofin and spermidine increased MMP (Δψm) loss. In addition, treatment with auranofin and spermidine increased the cleaved form of PARP cleaved by activated caspase-3 (Cohen, 1997; Luo & Kraus, 2012), a marker of apoptosis. Thus, our findings suggest that combined treatment with auranofin and spermidine could activate the mitochondrial apoptotic pathway. An elevated ROS level is a critical mechanism that induces cancer cell apoptosis. ROS overproduction, owing to impaired regulation of ROS homeostasis, has been examined as an upstream regulator of apoptosis (Gao et al., 2020). Therefore, we examined the relationship between ROS overproduction and mitochondria and determined the presence or absence of mitochondrial ROS generation following combined treatment with auranofin and spermidine. Based on the flow cytometry results and fluorescence imaging using H2DCF-DA, combined treatment with auranofin and spermidine led to ROS overproduction with colocalization of ROS and mitochondria. Moreover, NAC, a ROS scavenger, completely reversed the apoptosis-associated changes induced by auranofin and spermidine.
Phosphorylated and activated Akt by PI3K can regulate several functions related to cell survival, cell cycle, and cell growth (Sun et al., 2021; Wen et al., 2019). The PI3K/Akt pathway, which participates in various mechanisms related to cell survival, has been explored as a target for drug development. Furthermore, apoptosis due to the auranofin-induced inhibition of PI3K/Akt signaling has been previously investigated. Our findings suggest that the phosphorylated PI3K and Akt were markedly reduced upon treatment with auranofin and spermidine, whereas the PI3K inhibitor enhanced apoptosis induced by auranofin and spermidine. Herein, we observed that apoptosis induced by auranofin and spermidine was related to the suppression of the PI3K/Akt signaling pathway. Furthermore, we used NAC and PI3K inhibitor, which suppress and aggravate apoptosis induced by co-treatment with auranofin and spermidine, respectively, to identify key regulators of ROS generation or the PI3K/Akt signaling pathway in auranofin and spermidine-induced apoptosis. Our results indicate that auranofin and spermidine-induced apoptosis and mitochondrial dysfunction were completely restored following treatment with respective inhibitors. Accordingly, auranofin and spermidine increase Hep3B cell apoptosis, with ROS acting as an upstream regulator.
In conclusion, our results demonstrate that combination treatment with auranofin and spermidine can synergistically induce apoptosis by suppressing the ROS-mediated PI3K/Akt signaling pathway. Furthermore, auranofin and spermidine induced intrinsic apoptosis through mitochondrial ROS generation and regulation of Bcl-2 and Bax expression. Therefore, as a potential strategy to overcome the limitations and side effects of HCC therapy, we propose a combination of low-dose auranofin and spermidine as a synergistic combination of low-dose auranofin and spermidine therapy.